This protocol has been developed by Joseph D. Card, Guillaume Adelmant, and Jarrod A. Marto of the Dana-Farber Cancer Institute Inc., Brigham and Women’s Hospital, and Harvard Medical School, MA, USA.
This note is not and should not be construed as an endorsement of any product.
Email: [email protected]
Web: https://martolab.dana-farber.org/
Purpose
Sera-Mag and Sera-Mag SpeedBeads provide efficient magnetic bead separation technology for diverse genomic and proteomic applications. Carboxylic groups on the surface of the beads permit easy covalent coupling of target biomolecules of interest, such as proteins and nucleic acids, using convenient carbodiimide chemistry. The large cauliflower-shaped surface paired with proprietary Sera‑Mag and SpeedBead chemistry provides a large surface area which offers excellent sensitivity and low non-specific binding (Fig 1). The unique combination of these features provides fast reaction kinetics to maximize sample retention and reduce the amount of beads required. Sera‑Mag SpeedBeads have a double layer of magnetite applied through the core shell design process, providing more rapid magnetic manipulation compared to Sera-Mag beads, particularly in high viscosity samples.
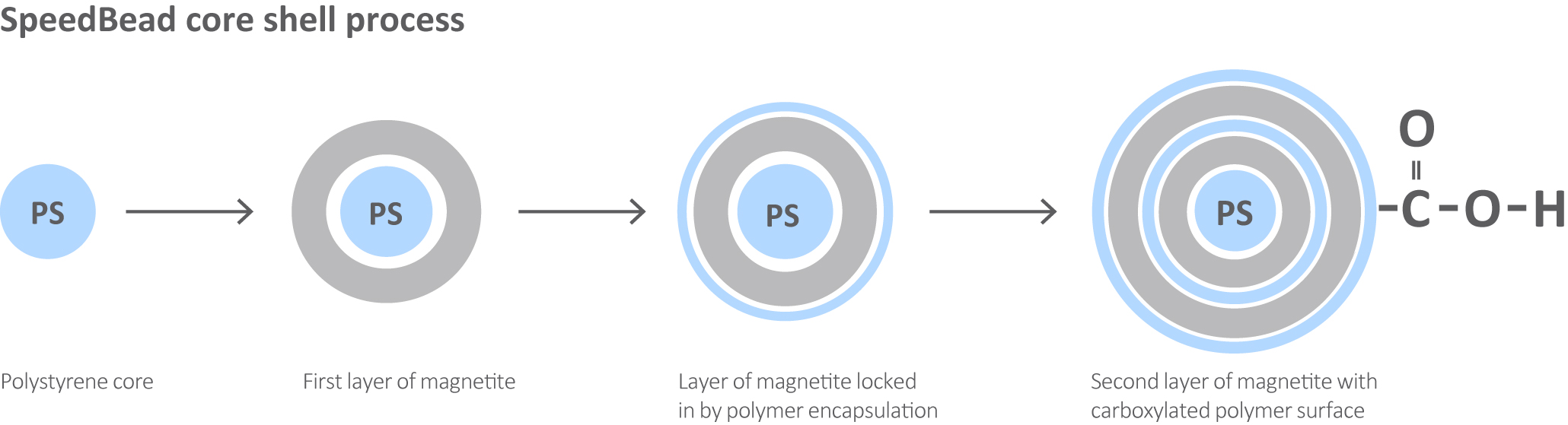
Fig 1. Sera-Mag and SpeedBead chemistry provides a large surface area which offers excellent sensitivity and low non-specific binding.
Find out more about Sera-Mag SpeedBeads
Protein and peptide single-pot solid-phase-enhanced sample preparation (SP3) with Sera-Mag SpeedBead Carboxylate-Modified Magnetic Particles is a novel technique that takes advantage of these features to enable the fast and efficient removal of a wide range of detergents, salts and other contaminants from protein and peptide samples prior to mass spectrometry analysis (1,2). As a result, SP3 provides a robust platform which is compatible with numerous workflows and sample types. As a scalable magnetic bead format, SP3 also provides significant cost and time savings over traditional sample cleanup methods such as batch-mode chromatography, solid-phase extraction and spin-filters. The protocol described in this technical note is applicable to inputs ranging from as few as 1000 sorted cells up to 20 mg of cell lysate (Fig 2).
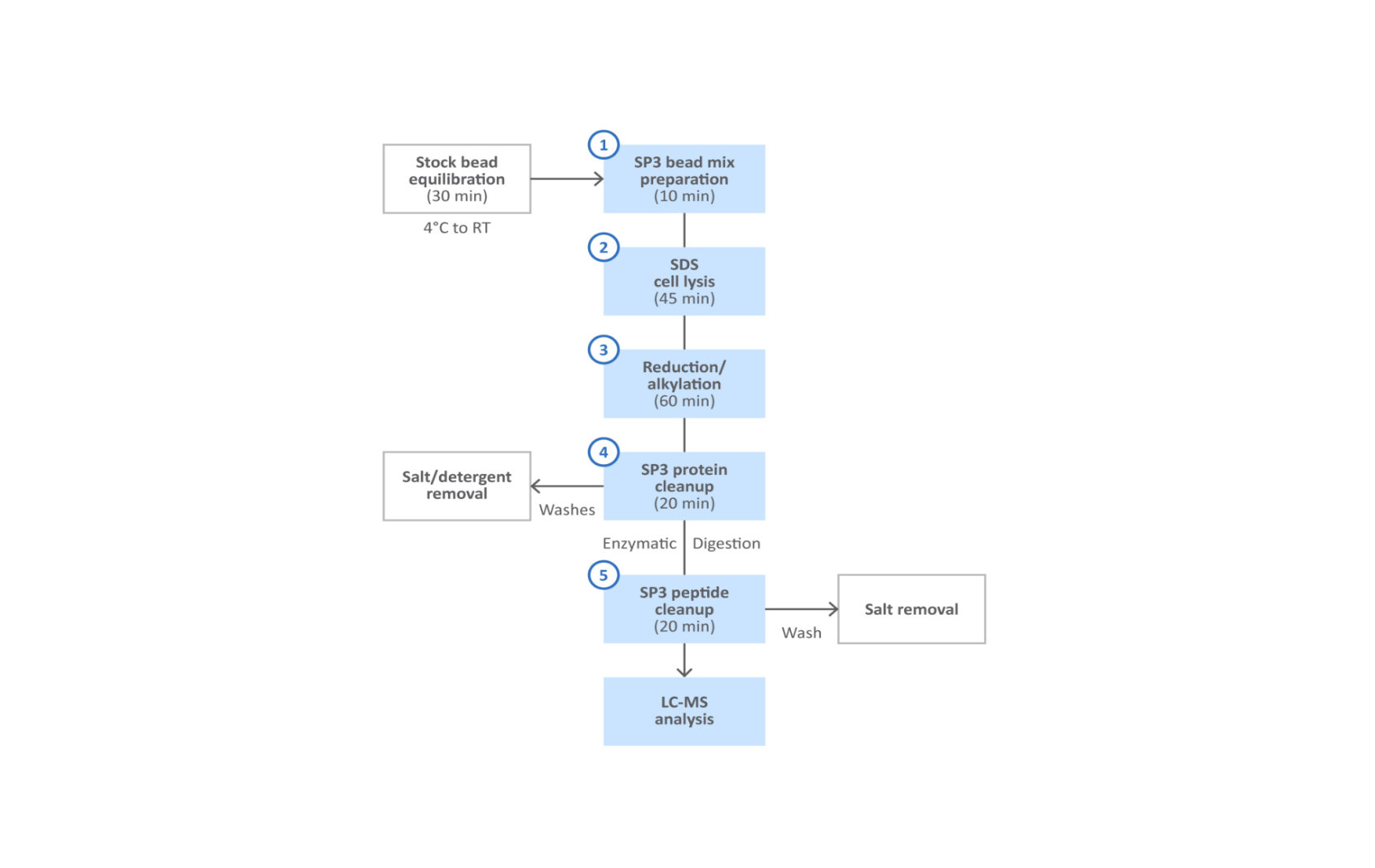
Fig 2. SP3 protein and peptide cleanup workflow.
Reagents and equipment
Lysis reagents
- PBS washed cell pellets.
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich Cat. #L6026) – prepare 10% stock solution and store at room temperature for up to 2 years.
- HEPES (Sigma-Aldrich Cat. #h2375) – prepare 1 M stock at pH 8.5 and store at 4°C for up to 2 years.
- Roche cOmplete™ Mini Protease Inhibitor Cocktail tablets (Sigma-Aldrich Cat. #11836153001).
- Benzonase nuclease ≥ 90% purity (Sigma-Aldrich Cat. #E1014).
- Dithiothreitol (DTT) (Sigma-Aldrich Cat. #D5545) – prepare 100 mM aliquots and store at -80°C for up to 1 year.
- Iodoacetamide (IAA) (Sigma-Aldrich Cat. #I1149) – prepare 550 mM aliquots and store at -80°C for up to 1 year.
SP3 protein cleanup and enzymatic digestion
- Sera-Mag Carboxylate SpeedBeads (Cat. #45152105050250 and Cat. #65152105050250).
- Ethanol (EtOH) HPLC/Spectrophotomeric grade 200 proof (Sigma-Aldrich Cat. #459828).
- Trypsin (Sequencing Grade, Promega Cat. #V5113).
- Ammonium Bicarbonate (AmBic) (Sigma Aldrich BioUltra - > 99.5% Cat. #09830) – prepare 100 mM buffer fresh.
- Ultrapure filtered water e.g. Barnstead Nanopure Diamond.
- Pierce™ BCA Protein Assay Kit Cat. #23227.
- SilverQuest™ silver staining kit (Life Technologies Cat. #LC6070).
- Bel-Art disposable pestles Cat. #199230001.
SP3 peptide cleanup
- Acetonitrile (MeCN) (Fisher Chemical Optima/LC-MS grade Cat. #A955).
- Dimethyl Sulfoxide (DMSO) (ACS Reagent ≥ 99.9% Sigma-Aldrich Cat. #472301).
- Pierce Quantitative Fluorometric Peptide Assay (Thermo Scientific Cat. #23290).
Equipment
- Magnetic stand such as the MagRack 6 for 1.5 mL microcentrifuge tubes.
- SP3 also works with other magnetic stand formats such as stands for 96 well plates or 15 or 50 mL conical tubes.
- Bath sonicator.
- Microcentrifuge tubes that demonstrate low binding of proteins and peptides and that can also tolerate high organic solvent concentrations without leaching plasticizers. Sarstedt™ 1.5 mL screw cap microcentrifuge tubes (Cat. #72.692) were used for all steps in this protocol.
- Temperature-controlled water bath or dry-heating block.
- Mixer platform (e.g. Eppendorf™ thermomixer).
- Centrifuge for pelleting cellular debris in the range of 16000 rcf e.g. Eppendorf 5415C or equivalent.
- Vacuum concentrator e.g. Thermo Scientific Savant™ SPD131DDA.
- pH paper strips e.g. MColorpHast™ pH 0–14 (MilliporeSigma Cat. #1095350001) for checking sample pH throughout the protocol.
Protocol
Important guidelines for SP3 compatibility (1)
- Buffers: SP3 works in a wide variety of buffers such as ammonium bicarbonate, HEPES and Tris-HCl below 1 M in concentration and in the pH range of 7 to 8.5.
- Detergents: SP3 protein and peptide clean-up is compatible with a wide range of detergents including SDS, NP-40 and RapiGest™. An SDS lysis buffer was used for data presented herein.
- Solvents: Protein binding to SP3 beads is promoted by increasing the concentration of organic solvents (such as ethanol or acetonitrile) above 50%. We use ethanol to perform protein-level purification in this protocol while SP3 peptide binding is performed in acetonitrile.
Nucleic acid shearing
- IMPORTANT: Genomic DNA released during lysis will coat the beads, causing them to aggregate or adhere to the tube walls or pipette tips. This behavior dramatically reduces protein binding and recovery. We recommend digesting nucleic acids with benzonase or shearing by mechanical methods such as sonication.
Bead concentration during binding
- We recommend maintaining a bead concentration of ≥ 0.5 μg/μL during protein binding and ≥ 0.1 μg/μL during peptide binding. We also recommend adding an excess of beads for sample inputs below 1 μg to maintain a visible bead volume. When scaling to the milligram range maintain the minimum of 0.5 μg/μL bead concentration while working in a larger volume sample tube.
Sample concentration
The binding capacity of SP3 beads provides a flexible clean-up format across a range of protein and peptide concentrations (10 μg/mL to 5 mg/mL), as long as the concentration of beads is adjusted as described above.
Sample handling
Throughout this protocol care should be taken to gently aspirate supernatant to avoid dislodging protein or peptide-bound beads from the magnet. 200 μL gel loading pipette tips for sample aspiration is recommended.
1. SP3 bead preparation
- 1.1 Sera-Mag Carboxylate SpeedBeads are stored at 50 mg/mL concentration (5% solids) in water with 0.05% sodium azide and should be stored at 4°C when not in use.
- 1.2 Take the beads out of storage at 4°C and let them equilibrate to room temperature for 30 minutes. If the beads have settled during storage they should be resuspended by inversion or gentle vortexing until no solid bead mass is visible at the bottom of the bottle. Preparing aliquots of stock beads avoids excess handling of the main bottles and minimizes the risk of contamination.
- 1.3 Prepare a fresh 1:1 bead mix before each experiment by combining equal volumes of stock beads in a clean Sarstedt tube.
- 1.4 Briefly vortex the mixture and place the tube on the magnetic stand for two minutes to collect the beads. Aspirate and discard the storage buffer.
- 1.5 Wash the beads with ultrapure water at a volume corresponding to 5–10× the initial volume of mixed beads.
- 1.6 Vortex the beads for 10 seconds and place on the magnetic stand for two minutes to collect the beads. Aspirate and discard the wash buffer.
- 1.7 Repeat the wash steps two times. 1.8 Resuspend the beads with ultrapure water at a final concentration of 10 μg/μL.
2. SDS cell lysis
- 2.1 SDS lysis buffer: Prepare fresh 50 mM HEPES pH 8.5/1% SDS from stock 1 M HEPES pH 8.5 and 10% SDS solutions. Add cOmplete Mini EDTA-free Protease Inhibitors (Roche) to 1× concentration.
- 2.2 Thaw cell pellets on ice.
- 2.3 Add four pellet volumes of SDS lysis buffer to the cell suspension. Homogenize using a disposable pestle (Bel-Art) with 20 strokes.
- 2.4 Add benzonase to a concentration of 500 units/mL and mix by inversion.
- 2.5 Incubate the lysate at 4°C for 30 minutes with end-over-end rotation.
- 2.5.1 Check the viscosity of the samples at this point by pipetting. If you are unable to pipette, add more benzonase and extend the incubation.
- 2.6 Pellet cellular debris by centrifugation at 16000 rcf for 10 minutes at 4°C and transfer the supernatant to a clean tube.
- 2.7 Quantify the protein content by BCA protein assay.
- 2.8 Normalize all samples to the same volume and concentration with SDS lysis buffer.
- 2.8.1 Aliquots of lysate can be frozen and stored at -80°C for later use.
3. Reduction and alkylation
- 3.1 Add DTT (100 mM stock) to a 10 mM final concentration and briefly vortex.
- 3.2 Reduce and denature the samples at 56°C for 30 minutes in a water bath.
- 3.3 Cool the samples to room temperature and centrifuge for 2 seconds to collect liquid from the sides of the tubes/caps.
- 3.4 Add IAA (550 mM stock) to a 22.5 mM final concentration and briefly vortex.
- 3.5 Alkylate the samples by incubating in the dark for 30 minutes at room temperature.
- 3.6 Add fresh 100 mM DTT to a final concentration of 10 mM to quench residual IAA and incubate for 10 minutes at room temperature.
4. SP3 protein cleanup
- 4.1 Check the pH of the sample is in the range of 7 to 8.5 for optimal binding by measuring an aliquot on pH paper.
- 4.1.1 Save a 1 μg aliquot of the lysate at this step and freeze it at -80°C for use as the “input” fraction for quality control by SDS-PAGE and silver staining.
- 4.2 Add washed beads (prepared as above) to the samples in a ratio of 5–10 μg of beads to 1 μg of protein and briefly vortex.
- 4.3 Immediately add a volume of 100% ethanol to the samples to obtain a 50% final concentration to initiate protein binding to the beads.
- 4.4 Vortex the samples to mix but ensure that beads are not stuck on the sides of the tube.
- 4.4.1 Note: The protein-bead mixture will be sticky at this stage. Avoid touching the beads to minimize sample loss.
- 4.5 Incubate the samples on a room temperature mixer platform for 10 minutes at 1000 rpm.
- 4.6 Remove the samples from the mixer, centrifuge them for two seconds and place them on the magnetic stand for two minutes.
- 4.7 Transfer the supernatants to a clean Sarstedt tube.
- 4.7.1 Save a 1 μg aliquot of supernatant as the “flow-through” fraction and freeze it at -80°C for quality control by SDS-PAGE and silver staining.
- 4.8 Wash the beads by adding a volume of 80% ethanol corresponding to at least twice the initial sample volume and vortex for 30 seconds.
- 4.9 Centrifuge the samples for two seconds and place back on the magnetic stand for one minute.
- 4.10 Remove the washes and save in a separate Sarstedt tube. Repeat two times.
- 4.10.1 Save a 1 μg aliquot of each wash fraction (“washes 1 to 3”) and freeze them at -80°C for quality control by SDS-PAGE and silver staining.
- 4.11 After the final wash, air dry the beads for 30 seconds to remove as much ethanol as possible.
- 4.12 Resuspend the beads in 100 mM ammonium bicarbonate buffer and vortex.
- 4.12.1 If the beads remain aggregated after vortexing, then briefly sonicate.
- 4.12.2 Save a 1 μg aliquot of bead slurry as the “bound” fraction and freeze it at -80°C for quality control analysis by SDS-PAGE and silver staining.
- 4.13 Add Promega™ sequencing grade trypsin in a 1:20 enzyme-tosubstrate ratio to each sample.
- 4.14 Ensure the pH for all samples is in the range of 7.5 to 8 and digest at 37°C overnight.
- 4.14.1 Digesting on a mixing platform or end-over-end rotator to keep the beads in suspension is recommended.
5. SP3 peptide cleanup
- 5.1 Note: Peptide binding to SP3 beads is promoted by the addition of acetonitrile to a final concentration ≥ 95%. The aqueous sample volume prior to dilution must not exceed the available tube volume (e.g. 50 μL of peptides would require the addition of 1 mL of 100% MeCN to reach ~95% MeCN concentration). If necessary, sample volume may be reduced by vacuum concentration prior to addition of MeCN. Complete drying to avoid potential sample loss is recommended against.
- 5.2 Remove the samples from incubation at 37°C and centrifuge for one minute at 2000 rcf to pellet the beads.
- 5.3 Incubate the digested samples on the magnetic stand for two minutes and transfer the supernatant to clean Sarstedt tubes.
- 5.3.1 Save an aliquot of the digested peptide supernatant for quality control analysis by fluorometric peptide assay and store at -80°C until needed.
- 5.4 Ensure the sample pH is still in the range of 7.5 to 8 after overnight digestion.
- 5.5 Add washed beads (prepared as above) to the samples in a ratio of 5–10 μg of beads to 1 μg of peptide and briefly vortex.
- 5.6 Immediately add a volume of 100% MeCN to reach ≥ 95% final concentration to initiate peptide binding to the beads.
- 5.7 Immediately vortex to maintain beads in suspension.
- 5.7.1 Concentrated peptide samples may cause the beads rapidly aggregate together or to the side of the tube after the addition of MeCN. If this occurs, briefly sonicate the tube and vortex until the beads are back in suspension.
- 5.8 Incubate all samples on a room temperature mixer for 10 minutes at 1000 rpm.
- 5.9 Remove the samples from the shaker and centrifuge for two seconds to collect any liquid or beads on the sides or cap.
- 5.10 Place the samples on the magnetic stand for two minutes.
- 5.11 Remove the supernatant and save in a clean Sarstedt tube. 5.11.1 Save an aliquot from the supernatant for analysis by fluorescent peptide assay and store at 80°C until needed.
- 5.12 Wash the beads with a volume of 100% MeCN corresponding to at least two times the initial sample volume and vortex for 30 seconds.
- 5.13 Centrifuge the samples for two seconds and place on the magnetic stand for one minute.
- 5.14 Aspirate the supernatant and save in a separate tube. 5.14.1 Save an aliquot of the wash for analysis by fluorometric peptide assay and store at -80°C until needed.
- 5.15 Air dry the beads for 30 seconds to ensure MeCN is removed before starting the elution.
- 5.16 Elute peptides with 2% DMSO in water in a volume equivalent to 5–10× the dried bead volume.
- 5.16.1 Add the elution buffer to the dried beads; avoid touching the beads with the pipette tip to avoid sample loss.
- 5.17 Sonicate the samples for one minute in a bath sonicator to disrupt any aggregated beads and to promote the release of bound peptides.
- 5.18 Incubate the samples on a room temperature mixer platform for five minutes at 1000 rpm.
- 5.19 Remove the samples from the mixer and centrifuge for two seconds.
- 5.20 Place them on the magnetic stand for two minutes.
- 5.21 Optional step to prevent bead carryover: aspirate the eluates as above and transfer to new tubes. Incubate on the magnetic stand for two minutes and transfer the supernatants to clean tubes.
- 5.21.1 Save an aliquot of the eluate for quality control analysis by fluorometric peptide assay and store at -80°C until needed.
- 5.22 Samples are ready for immediate analysis by LC-MS/MS or storage at -80°C.
Quality controls
Silver stain for protein level SP3
- Goal: analyze each fraction by SDS-PAGE and silver staining to assess SP3 protein binding efficiency.
- Dilute the 1 μg “input” aliquot (set aside in step 4.1) to 20 μL with loading buffer to 1× final concentration.
- Dry the “flow-through” and “wash” aliquots (saved in steps 4.7 and 4.10) to completion in a vacuum concentrator to remove all ethanol from the samples prior to loading on the gel. Resolubilize the dried protein in 20 μL 1× loading buffer.
- Dilute the 1 μg “bound” aliquot to 20 μL with loading buffer to 1× final concentration.
- Denature all samples at 95°C for 10 minutes.
- Place the “bound” aliquots (step 4.12) on the magnetic stand for two minutes to collect the beads.
- Load 10 μL of each sample on the gel (equivalent to 0.5 μg).
- Run the gel and perform silver staining according to the manufacturer’s instructions.
- Visually compare protein bands across the lanes for the input and bound fractions to assess binding efficiency. Proteins present in the flow-through or wash fractions may result from using incorrect organic solvent concentration, from not using enough beads during the binding step or from bead carry-over.
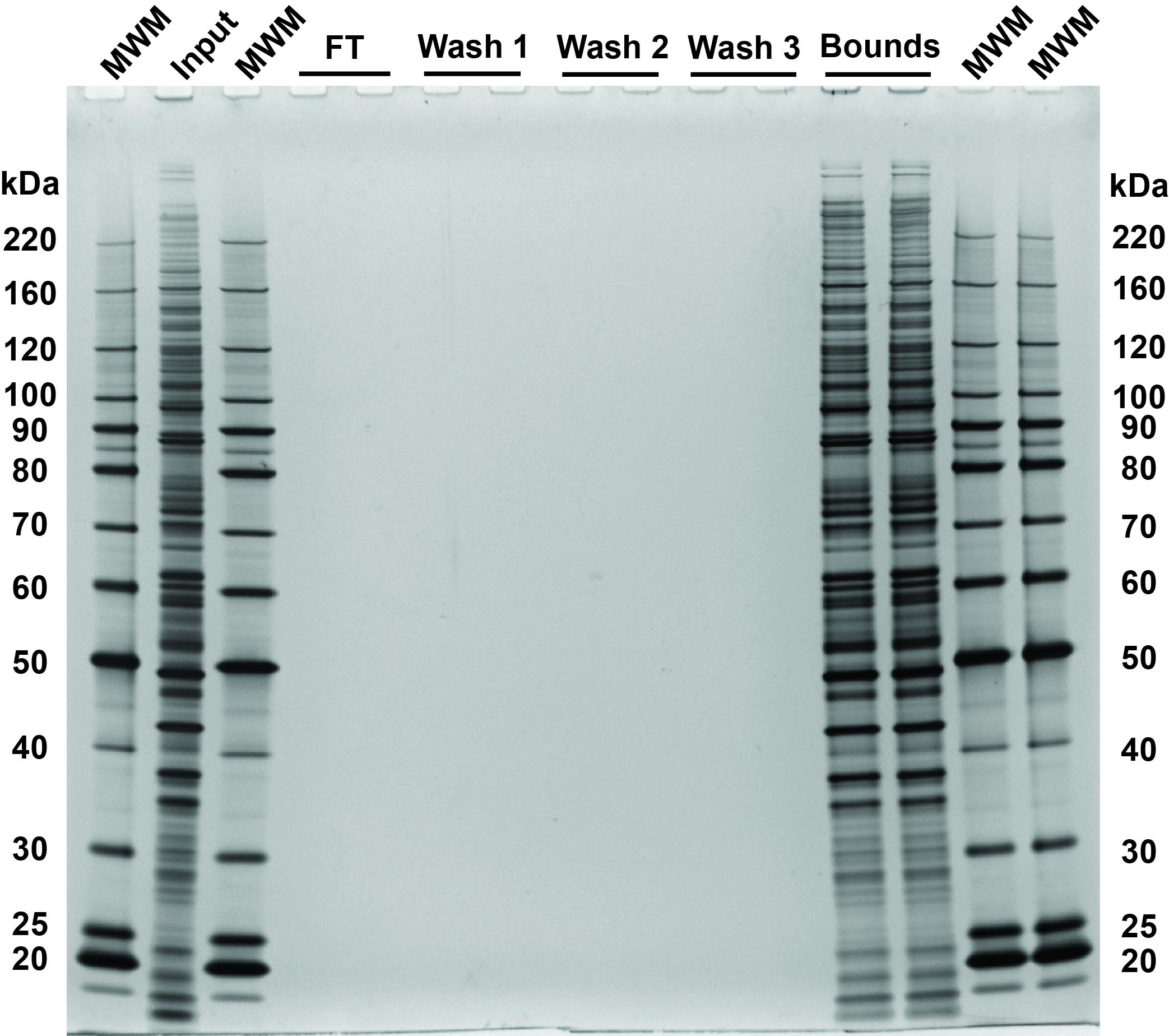
Fig 3. SDS-PAGE and silver stain visualization of 10 μg replicate SP3 protein cleanups. Comparison of input and bound fractions illustrates effectiveness of SP3 protein-level protocol. HeLa S3 cells were lysed in SDS, treated with benzonase, reduced and alkylated according to the protocol in sections 2–3. SP3 cleanup was performed according to the protocol in section 4. Aliquots corresponding to 1 μg of each fraction were denatured at 95°C for 10 minutes, with 0.5 μg of each fraction loaded on the gel.
Fluorometric peptide assay for peptide SP3 cleanup
- Goal: quantify the peptide content of each SP3 peptide fraction to assess binding efficiency, sample recovery, or general troubleshooting.
- Dry the digested peptide supernatant, flow-through, wash, and elution aliquots (set aside as described above) with a vacuum concentrator to remove any MeCN.
- Reconstitute peptides in water and follow assay instructions.
- If peptides are detected in the flow through and/or wash they can be recovered by combining the saved supernatant and wash and drying them down in a vacuum concentrator. Reconstitute the dried sample in 100 mM ammonium bicarbonate pH 8, add fresh beads and repeat the peptide cleanup process.
Mass spectrometry
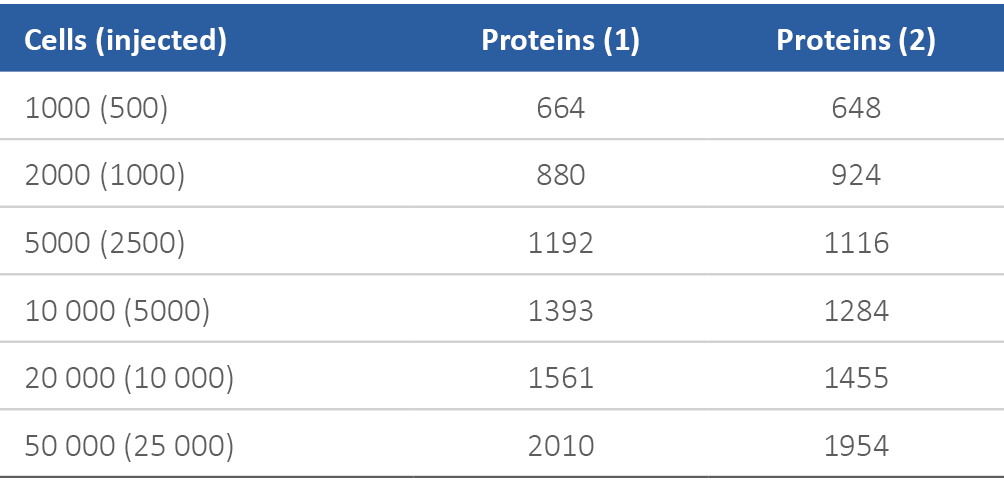
FACS sorted cells were collected in a 96-well plate as indicated in Table 1. The lysis buffer was composed of 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1.25% SDS with EDTA-free protease inhibitors. Sorted cells were kept on ice while 80 μL of lysis buffer was added to each well, mixed by pipetting 3× and transferred to clean tubes. Benzonase treatment was modified to use 10 units/mL for lysates derived from 1000–20000 cells and 25 units/mL for 50000 cells. Reduction and alkylation were performed as described in section 3. Protein and peptide level SP3 cleanups were performed as described in sections 4 and 5 with 100 μg of fresh beads for each cleanup.
Table 1. Reproducible analysis of FACS sorted cells with SP3 and LC-MS/MS.
LC -MS/MS analysis was performed by injecting half of each eluate onto a precolumn (100 μm I.D. × 4 cm POROS 10R2, Applied Biosystems) coupled directly to a self-packed analytical column (30 μm I.D × 50 cm Monitor C18, Column Engineering), followed by gradient elution (Waters NanoAcquity™ UHPLC system; 2%–35% B in 45 minutes; A = 0.2 M acetic acid in water, B = 0.2 M acetic acid in acetonitrile) into the mass spectrometer (Q Exactive™ HF mass spectrometer, Thermo, Waltham, MA) equipped with a Digital PicoView electrospray source platform (New Objective, Woburn, MA) (3). The spectrometer was operated in data dependent mode where the top 10 most abundant ions in each MS scan were subjected to MS/MS in the HCD cell with normalized collision energy = 27. MS spectra were recalibrated using the background ion (Si(Ch2)2O)6 at m/z 445.12 +/- 0.03 and converted into a Mascot™ generic file format (.mgf) using multiplierz scripts (4). Spectra were searched using Mascot (version 2.6). Peptide spectral matches (PSM) to the reverse database were used to calculate a global false discovery rate and were discarded. Data were further processed to remove PSMs with an FDR greater than 1.0%.
Large scale SP3 guidelines
Protein level SP3 scales well to work with large amounts of input by increasing the volume of the sample container (e.g. 15 mL conical vial), magnetic stand size and the volumes used during each step while maintaining the bead concentration of 0.5 μg/μL. When working in the milligram range a large aggregate of beads and protein will form after the addition of ethanol. Due to the volume of this aggregate extending the time for the protein binding step and for all subsequent incubations of the beads on the magnetic stand is recommended. The aggregate volume can be reduced further by diluting the protein concentration to 1–2 mg/mL although that will require the addition of more beads to maintain the minimum concentration. Increasing the wash volume to at least 5× the initial sample volume to effectively remove lysis buffer reagents is also recommended. Vacuum concentration for removing any residual ethanol after aspirating the supernatant from the final wash when working at larger scales is recommended. Avoid over-drying the beads otherwise they will be difficult to resuspend in the digestion buffer. Digesting in a volume sufficient to keep the protein concentration below 4 mg/mL to avoid protein precipitation is recommended. Trypsin can be scaled down to an enzyme/substrate ratio of 1:50 if enzyme cost is a concern.
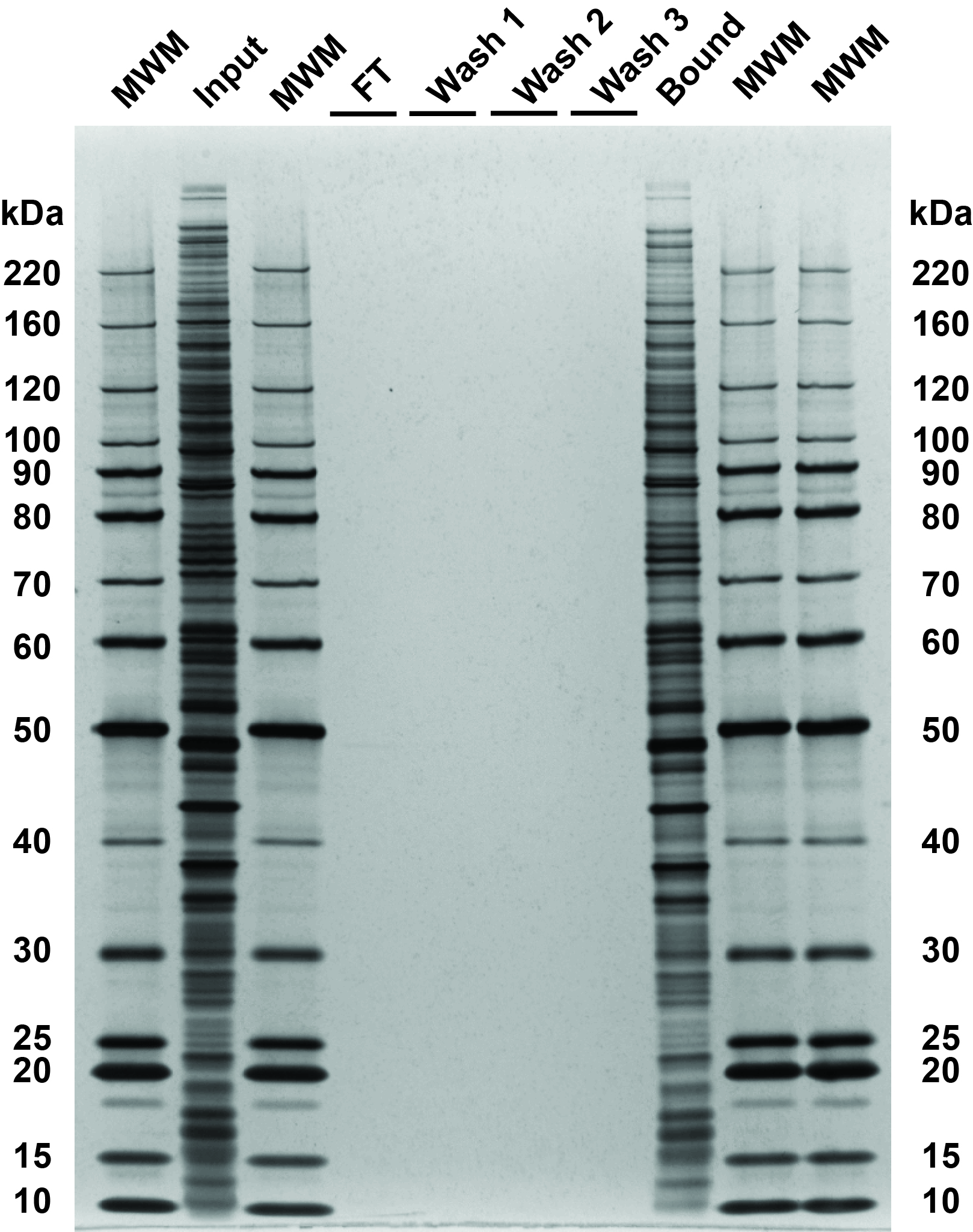
Fig 4. SDS-PAGE and silver stain visualization of SP3 protein cleanup of 20 mg HeLa S3 lysate. Comparison of input and bound fractions illustrates effectiveness of SP3 protocol for larger protein input. HeLa S3 cells were lysed in SDS-lysis buffer, digested with benzonase and reduced and alkylated according to the protocol in sections 2–4. The protein binding time and incubation on the magnetic stand were both extended to 20 minutes. The bound protein was washed with 3× 10 mL of 80% ethanol (5× the initial protein volume) by vortexing for 30 seconds at maximum speed followed by incubation on the magnetic stand for five minutes after each wash. 20 μg aliquots were taken at all steps for silver staining with 0.5 μg of each fraction loaded on the gel following the quality control protocol in section 6. The large scale SP3 protein cleanup procedure for 20 mg visualized in Figure 4 demonstrates quantitative recovery of protein comparable to that achieved at the 10 μg scale in Figure 3.
Discover our broad portfolio of magnetic beads- Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, Krijgsveld J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Sys. Biol. 2014 Oct 30;10:757. doi: 10.15252/msb.20145625.
- Hughes CS, Moggridge S, Müller T, Sorensen PH, Morin GB, Krijgsveld. Single-pot, solid-phase enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019 Jan;14(1):68‒85. doi: 10.1038/s41596-018-0082-x.
- Ficarro SB1, Zhang Y, Lu Y, Moghimi AR, Askenazi M, Hyatt E, Smith ED, et al. Improved electrospray ionization efficiency compensates for diminished chromatographic resolution and enables proteomics analysis of tyrosine signaling in embryonic stem cells. Anal. Chem. 2009 May 1;81(9):3440‒7. doi: 10.1021/ac802720e.
- Alexander WM, Ficarro SB, Adelmant G, Marto JA. multiplierz v2.0: A Python-based ecosystem for shared access and analysis of native mass spectrometry data. Proteomics. 2017 Aug;17(15‒16). doi: 10.1002/pmic.201700091.