Cryo electron microscopy (cryo-EM) provides possibilities to determine structures for more challenging proteins, such as membrane proteins and larger protein complexes. This blog provides tips for preparing cryo-EM samples.
Knowing the three-dimensional structure is essential in understanding the function of a protein. In structural biology, X-ray crystallography is well established to determine the three-dimensional structure of proteins.
Cryo-EM provides possibilities to determine structures for more challenging proteins, such as membrane proteins and larger protein complexes which were previously too difficult to crystallize or too large to study with NMR. The past years of technical improvements have made cryo-EM a very important and widely used technique within structural biology. The number of structures determined using cryo-EM are increasing rapidly.
The cryo-EM workflow starts with cloning and expression of the target which is then purified and characterized before the sample is applied on grids and analyzed with cryo-EM (Fig 1).
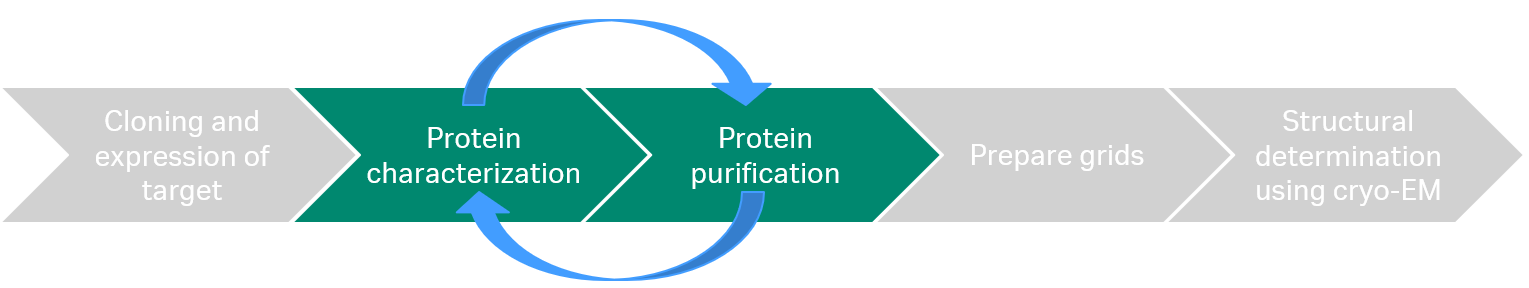
Fig 1. The cryo-EM workflow: After cloning and expression, the target protein is purified and characterized. Finally, the sample is applied on grids and analyzed with cryo-EM.
In our webinar, we focused on the protein characterization and protein purification, which often requires an iterative process to reach the required sample quality for cryo-EM to be successful.
Watch our webinar
Questions and answers
During our live webinar, questions were received from the audience and answered by our experts live. Below you can find these questions with responses.
Lotta Hedkvist has responded to general questions related to cryo-EM.

Lotta Hedkvist
Global Product Manager, ÄKTA systems at Cytiva
Lotta provides excellent chromatography system solutions to support scientists globally.
What is the biggest challenge when preparing samples for cryo-EM?
Preparing the grids is a big hurdle. A lot of time can be spent on testing different buffers and detergents to optimize vitrification to achieve a thin layer of amorphous ice with the protein sample embedded in random orientations. This can take from days and up to several months to optimize.
What factors do you need to consider for a successful cryo-EM experiment?
Ensuring homogeneity; both composition and conformation of the sample is important. Compositional homogeneity can be tested using SDS-PAGE, mass spectrometry, size exclusion chromatography, and dynamic light scattering (DLS). Conformational homogeneity is verified with negative staining and transmission electron microscopy (TEM). Characterization by biochemical and biophysical techniques (such as surface plasmon resonance, SPR) if sample amount allows can provide additional useful information.
What's the lower end limit on protein size for cryo-EM?
A size limit around a molecular weight of 50 000 in size is mentioned in the literature.
Veronica Fridh has provided insights to characterization using Biacore analysis.

Veronica Fridh
Global Product Manager, Biacore systems at Cytiva
Veronica helps customers obtain high-quality molecular interaction data as easily as possible.
How much sample do I need for a Biacore surface plasmon resonance experiment?
It depends on the affinity of the interaction and the size of your samples. Roughly, around 100 µL at 0.1 mg/mL for a first experiment. In comparison to cryo-EM, it generally means a higher volume but at a lower concentration.
Can I study membrane proteins by surface plasmon resonance? Amphiphols?
Yes, membrane proteins have been successfully studied in Biacore systems using different approaches such as detergent solubilization and with membrane-like nanodiscs. If amphipols are used as an alternative to detergents for solubilization, the easiest way to use them in Biacore would be to utilize a biotin-tag and capture them on Sensor Chip SA.
If you perform a Biacore experiment on the interaction of a large and a small protein, which is better to immobilize? The large or the small protein?
If there is a significant difference in the molecular weight of your proteins, attach the smaller protein molecule. There are also other aspects to consider: for the easiest approach, attach the protein with the available tag. To save on sample, I recommended you attach the sample available in the lowest amount. If your sample preparations are of different purity, it is better to put the purest sample on the sensor chip.
Why would you choose Biacore over other techniques for your analysis, such as MST or ITC?
Surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), and microscale thermophoresis (MST) are all label-free technologies. You can use them for quantitation of molecular interactions without interference from radioisotopes or fluorescently labeled ligands. Biacore SPR has been used for 30 years and appears in more than > 55 000 publications. Biacore SPR is often the first choice when it comes to the wealth of data generated, reliability, sensitivity, dynamic range, and specific applications such as binding kinetics. As for all techniques, I recommend that you use/verify your data with an orthogonal technique if in any doubt.
How do I know which protein I should put on the sensor chip and which to flow over?
The easiest approach is to attach the protein with the available tag. To save on sample, attach the sample available in the lowest amount. If your sample preparations are of different purity, it is better to put the purest sample on the sensor chip. If there is a significant difference in the molecular weight of your proteins, attach the smaller protein molecule.
How does knowing the affinity of a complex help with knowing what protein concentration to use?
By knowing the affinity, you know the equilibrium concentration of your binding partners, that is, when half of the complex is formed and half is not. For a robust measurement of Kd, you should use sample concentrations of up to double the concentration as the affinity. If you determine the affinity from binding kinetics as kd/ka, we recommend use of sample concentrations in the range of 0.1 to 10 or 100 × KD.
Below you will find questions about purifying proteins for cryo-EM preps, answered by Emma Lind.

Emma Lind
Global Product Manager, resins at Cytiva
Emma develops innovative chromatography solutions for research and process development.
In the presentation slides about purifications, the sequence of protein purification was from affinity chromatography to ion exchange to size exclusion chromatography (marked by arrows from left to right). Does this sequence have to be followed? Why can’t an affinity column be used as the last step?
How many steps you perform depends on the purity that you need for your sample. For cryo-EM, use of an affinity tag step followed by size exclusion chromatography often works. This approach means you can eliminate the ion exchange chromatography step, but it depends on the sample. You can find out more in this blog post on combining chromatography techniques.
Since only around up to 5 mg of protein is needed for cryo-EM, is it better to use affinity tags like FLAG™ or Rho1D4 that result in less protein, but in higher purity? Could the ion-exchange step then be skipped?
See my response to the previous question.
Which protein tags are suitable for cryo-EM sample proteins?
Cryo-EM should not restrict your tag usage. As with any tag purification, we suggest that you start with a His-tag. If that does not work, you can try a Strep-tag™ II for better purity, or GST for improved solubility.
Visit cytiva.com for more information on tags.
How is your Ni Sepharose excel resin different from your other Ni resins? Is it affected by detergents?
Ni Sepharose excel is an immobilized metal ion affinity chromatography (IMAC) resin, precharged with nickel ions. The resin is designed mainly for capture and purification of histidine-tagged proteins secreted into eukaryotic cell culture supernatants.
The very strong binding of nickel ions to Ni Sepharose excel enables direct loading of large sample volumes. This can be achieved without having to remove agents that could cause stripping of nickel ions from conventional IMAC resins. The strong nickel ion binding also provides very high resistance to EDTA and reducing agents like DTT.
Ni Sepharose excel is also suitable for purification of histidine-tagged proteins from other samples, including E. coli lysates.
There is currently no data available for how Ni Sepharose excel resin is affected by detergents.
RESOURCE Q would be also considered a good column in terms of resolution as well as Capto HiRes Q? What are the differences between them?
Our RESOURCE Q and RESOURCE S columns contain SOURCE 15 resins composed of 15 µm beads. Our Capto HiRes resin is composed of 9 µm Capto beads, which will give higher resolution. RESOURCE columns are PEEK columns and can be run at faster flow rates while our Capto HiRes columns are glass and give better resolution, which can be a benefit for samples for cryo-EM.
Is there any difference in Mono Q/Mono S resin and Capto HiRes Q/ Capto HiRes S resin?
Our Capto HiRes Q and S columns replace Mono Q and S, which we will discontinue due to chemical regulations. Our Capto HiRes columns offer some increase in resolution and separation compared to the Mono Q and S columns.
Do you just have the Superdex 6 Increase size exclusion column for protein complexes, or do you have resins for other proteins also?
Cytiva offers a range of size exclusion chromatography resins, for purification of small peptides up to large biomolecules and protein complexes. Within our Increase range we have:
- Superdex 200 Increase: Mr ~ 10 000 to ~ 600 000 for molecules such as antibodies
- Superose 6 Increase: Mr ~ 5000 to ~ 5 000 000 for large molecules such as protein complexes
- Superdex 75 Increase: Mr ~ 3000 to ~ 70 000 that can be used with most recombinant tagged proteins
- Superdex 30 Increase: Mr ~ 100 to ~ 7000 that is suitable for peptides and other small biomolecules.
To select the right size exclusion chromatography resin visit the Purify app.
What size exclusion chromatography column (a.k.a. gel filtration) would you advise to use for large membrane proteins?
Detergents are often expensive, and it is therefore useful to limit consumption for cost reasons. Small columns require less buffer therefore working with small sample volumes and the columns 3.2/300 and 5/150 columns are suggested when working with membrane proteins.
Select the size exclusion chromatography resin based on the size of your protein, see fractionation ranges listed in the question above.
Which column would you recommend for highest resolution: a Superdex 200 Increase 3.2/300, 10/300, or 5/150?
Select the column based on sample volume and what you plan to use your purified sample for. Longer columns provide higher resolution, therefore Superdex 200 Increase 3.2/300 or Superdex 200 Increase 10/300 is recommended for high resolution.
What options exist to remove additives like glycerol from a sample?
If you do need to buffer exchange, you can use our HiTrap Desalting columns and PD 10 columns containing Sephadex 25. Since the glycerol makes the sample viscous, you will probably need to decrease your flow rate to ensure a good separation.
Which detergents work with your resins?
Cytiva resins can be used with a wide range of detergents and reagents. Resins such as our Ni Sepharose Excel resin can be used to purify His-tagged proteins and are resistant to EDTA and reducing agents like DTT.
A detergent screening process takes time and resources that a small lab may not have. What can you tell me about nanodiscs as an alternative to detergents to stabilize membrane proteins outside the cell membrane? Are these suitable for cryo-EM?
Nanodiscs are, as you mention, an alternative to the use of detergents. There are some publications showing examples of the use of nanodiscs in EM studies (e.g., Denisov and Sligar, 2016).
Detergents need screening, but do you have a favorite one that you always start with?
Which detergent to use is determined by your protein and its stability in that detergent. Screening is good to ensure that yield/purity and capacity are maintained.
Is there a solution for purification of RNA binding proteins, which have intrinsically disordered domains?
Purifying such protein complexes via size exclusion chromatography is a challenge. However, other chromatography techniques (e.g., high-resolution ion exchange chromatography) might give an alternative to achieve your needed purity. In such cases, you can try to use a desalting column in the end to transfer your target into the right buffer.
Do you suggest in vivo RNA and protein complex purification or purify protein at first step and further add RNA molecule to make it a stable complex?
If both components (RNA and protein complex) are stable on their own and form complexes in vitro, purifying the individual components will enable you to perform a wider range of in vitro studies. Purifying the native full RNA-protein complex will give you deeper insights into the in vivo situation, so it depends on what you want to achieve.
Martin Sichting provided answers to questions about how the Micro kit for ÄKTA pure 25 M is used for microscale purification.

Martin Sichting
Application Developer at Cytiva
Martin helps users to get the best from their protein purification.
How long does it take to convert my ÄKTA pure 25 M with the Micro kit and how is this done? Is it practical to change back and forth between using the Micro kit and standard flow path? It seems a little impractical to dedicate an entire ÄKTA pure unit to cryo-EM prep alone. Can we leave the Micro kit hooked up to the ÄKTA and continue to use the regular functionality?
The replumbing takes about 10 to 15 min. You can do it yourself, following a detailed instruction provided with the Micro kit. We also offer the support from our service team for installation of the Micro kit. To utilize the full flow rate specification of the standard ÄKTA pure 25 M, you need to convert back again.
How does the thin tubing affect the overall pressure of the system?
When looking at the same volumetric flow rates (mL/min), thinner tubing will generate higher back pressure. High-resolution columns commonly used in micropurification are either more pressure stable (in case of IEX) than the columns used for preparative purification and/or run at lower flow rates (in case of SEC). Therefore, this increase in pressure does not harm your application.
Can you still use the Micro kit for other purification that uses a fast flow (like ion exchange chromatography)? Will the pressure rapidly increase with the smaller tubing?
Depending on the column you want to use, the Micro kit may not allow maximum flow rate (e.g., when using a 5 mL HiTrap column). You can then change the setup of your ÄKTA pure from micropurification to preparative purification. As mentioned above, it takes around 10 to 15 min to convert the flow path and components.
With the low flow rates used for the Micro kit on ÄKTA pure 25 M, do I need to take extra precautions such as extensive eluent degassing to ensure flow accuracy?
Cytiva recommends filtering and degassing buffers for the use on ÄKTA pure systems whether you use the Micro kit or not. There are no further precautions (e.g., online degassers) needed to operate the Micro kit.
In combination with the Micro kit, what is the recommended fraction collector?
The Micro kit contains a micronozzle for the Fraction collector F9-R. For collection in plates, this is currently not available but stay tuned for future updates.
Do you need special injection loops with the Micro kit?
No, you can use the loops that are used for the standard ÄKTA pure.
Is the column valve to be bypassed when connecting the Micro kit to ÄKTA pure 25 M?
Each valve that you add to the flow path will cause peak broadening and therefore the column valve is not recommended to be used with the Micro kit.
Is there any software update needed to operate the ÄKTA pure with the Micro kit. If so, what updates and do you charge for them?
The Micro kit for ÄKTA pure works with ÄKTA pure Instrument Configuration software 1.13 and later versions. It also works with UNICORN 6.3.2 or later versions. You can download the instrument configuration from the ÄKTA pure webpage free of charge.
When using the Micro kit, can I still use the overall total amount of protein or do I need to down scale the total amount of protein that is purified on a normal ÄKTA pure setup?
The amount of protein you can use depends on the capacity of the column you want to use. When using very high protein concentrations in SEC you might also need to consider increasing viscosity. The use of the Micro kit per se does not limit you in the protein amount used.
What is the sample loss percentage with the Micro kit with size exclusion chromatography (e.g., on a 3.2/300 column) when collecting fractionated samples?
Sample loss can occur at lot of different stages of your purification (transfer of sample, emptying the sample loop, unspecific interactions with your column, etc.). So, to answer this question is very challenging. When you have your setup under good control, I would expect a 5% sample loss.
What is the difference between an SEC run on an ÄKTA pure system with the small peak tubing with and without the Micro kit? Do you have a plot to show the difference?
Please refer to slide number 30 in the webinar presentation for this data.
Is it possible to extract bovine serum with ÄKTA systems?
You can use our Blue Sepharose for this purpose.