The FDA requires biopharmaceutical manufacturers to understand and control sources of variation according to the risks they present to process and product. Cell culture media and feeds have been shown to affect cellular performance, product quality, or both. The evolution of media formulas to those that are serum free and chemically defined poses both a challenge and an opportunity. The challenge is identifying which of the many components have the greatest impact on cells and the molecules they produce. The opportunity comes from the ability to uncover potential sources of variation previously masked by serum or other undefined additives, such as hydrolysates and lipid complexes. Advances in analytical methods, paired with effective, risk-based assessment methods, enhance understanding and help mitigate risks, enabling management of variation to the highest extent possible.
Raw material control requires understanding
A deep understanding of a product and its process are key to successful biopharmaceutical development. According to ICH Q8(R2), identifying sources of variability is another important aspect (1). The FDA’s guidance on process validation discusses variation in more detail, including the needs to understand sources, characterize variability, understand process impacts, and control variation using a risk-based approach (2). Ideally, 100 percent of the variation will be understood and accounted for, regardless of whether it has a positive or negative impact.
Variability can be introduced to biomanufacturing processes from many sources. Among these are human factors, equipment, measurement methods, process controls, and materials (3). The last category is especially relevant for cell culture media and feeds because variations at this stage can influence cell growth, productivity, and, ultimately, the quality of the molecule the cells produce.
Variability in materials used to make media and feeds can impact critical quality attributes (CQAs) of recombinant proteins in numerous ways. A few of the potential effects on monoclonal antibodies and vaccines are aggregation, amino acid substitution, truncation, and glycosylation changes (4). These variations can affect product potency, clearance, and/or safety.
Rising significance of impurities
Traditionally, mammalian cell culture media were basal formulas supplemented with bovine sera. Sera contain thousands of components, which can bring substantial variation to a biomanufacturing process. Also, because sera are animal-derived, they carry a risk of introducing adventitious agents, such as viruses. To address these challenges, the industry has moved away from using serum, replacing it with rich chemically defined (CD) media. CD formulations are highly complex, some with more than 120 individual components. Trace elements, major ions, amino acids, and vitamins are just a few of the types of ingredients used in combination to support cell growth and productivity.
Several factors help the industry be more aware of variation and understand where it comes from. First, the transition to more chemically defined media has led to a heightened awareness of small variations in raw materials. This is true because many of these modern media do not include sera or other undefined constituents. Since sera are inherently variable and contain thousands of components, such small variations in other raw materials are easily masked or obscured. In addition, the effects of small variations are amplified with the higher productivities achieved in today’s cell cultures. Lastly, analytical methods have evolved to provide better tools and higher sensitivity for understanding material variations.
Raw Materials Summits were held in the past two years, the first hosted by Amgen and the second hosted by Cytiva. Meeting attendees are global drug substance manufacturers and n-1 and n-2 suppliers. The goal is to bring a common understanding of the issues around raw material quality and the subsequent impacts to biomanufacturing products and processes. Many examples of the impact of raw material variation are presented at the summit. A summary of the 2018 content can be found on our website.
Risk assessment of raw materials
The purity of raw materials for media and feeds varies widely, depending on the supplier and specified grade. Cell culture grade materials are typically more than 99 percent pure (the same purity required by USP and ACS). Analytical grade materials have slightly higher purity, generally more than 99.5 percent. Although 99.5 percent sounds high, this means that one part per 200 represents one or more impurities.
Impurities affect cell cultures and product in different ways. Some are neutral, but others will have a positive or negative impact. Even impurities with a positive effect are undesirable if they cannot be understood, measured, and controlled to some degree. Characterization is required to understand what these impurities are, where they are coming from, and how to mitigate the associated risks.
Using appropriate analytical methods, impurities can be detected and their levels quantified. This characterization alone allows the identification of higher-quality lots to be sourced where possible. However, lot selection is not an ideal outcome from this activity and should be used only in cases where a more strategic option is not available. Determining the degree of variation in materials allows implementation of risk-based processes to focus on problematic materials and the ability to identify purer primary sources or processes. Combining material characterization and understanding with the trend toward increased supply chain transparency provides strategic opportunities for both manufacturers and suppliers to identify and implement improvements in raw material quality.
Understanding raw material supply chains
The supply chain for cell culture materials in bioprocessing can be very complex. The chart shown in Figure 1 depicts a simplified supply chain model. In this case, drug substance manufacturers (n) source raw material in the form of cell culture media and feeds from suppliers (n-1). Cell culture media suppliers source their raw material in the form of cell culture media constituents from chemical suppliers (n-2). Chemical suppliers then source their raw materials from natural resource extractive, reclamation, and synthesis industries (n-3). Often, however, the supply chain is much more complex, with drug substance manufacturers (n) sometimes also acting as n-1 or even n-2 suppliers. Furthermore, suppliers of cell culture media and feeds (n-1) can also be chemical suppliers (n-2), who may merely be distributing the chemicals that they source from other chemical suppliers (n-2). In some instances, this might be just repackaging and/or relabeling, which can obscure important information around material origin. This complexity increases the need for digital data exchange and supply chain transparency.
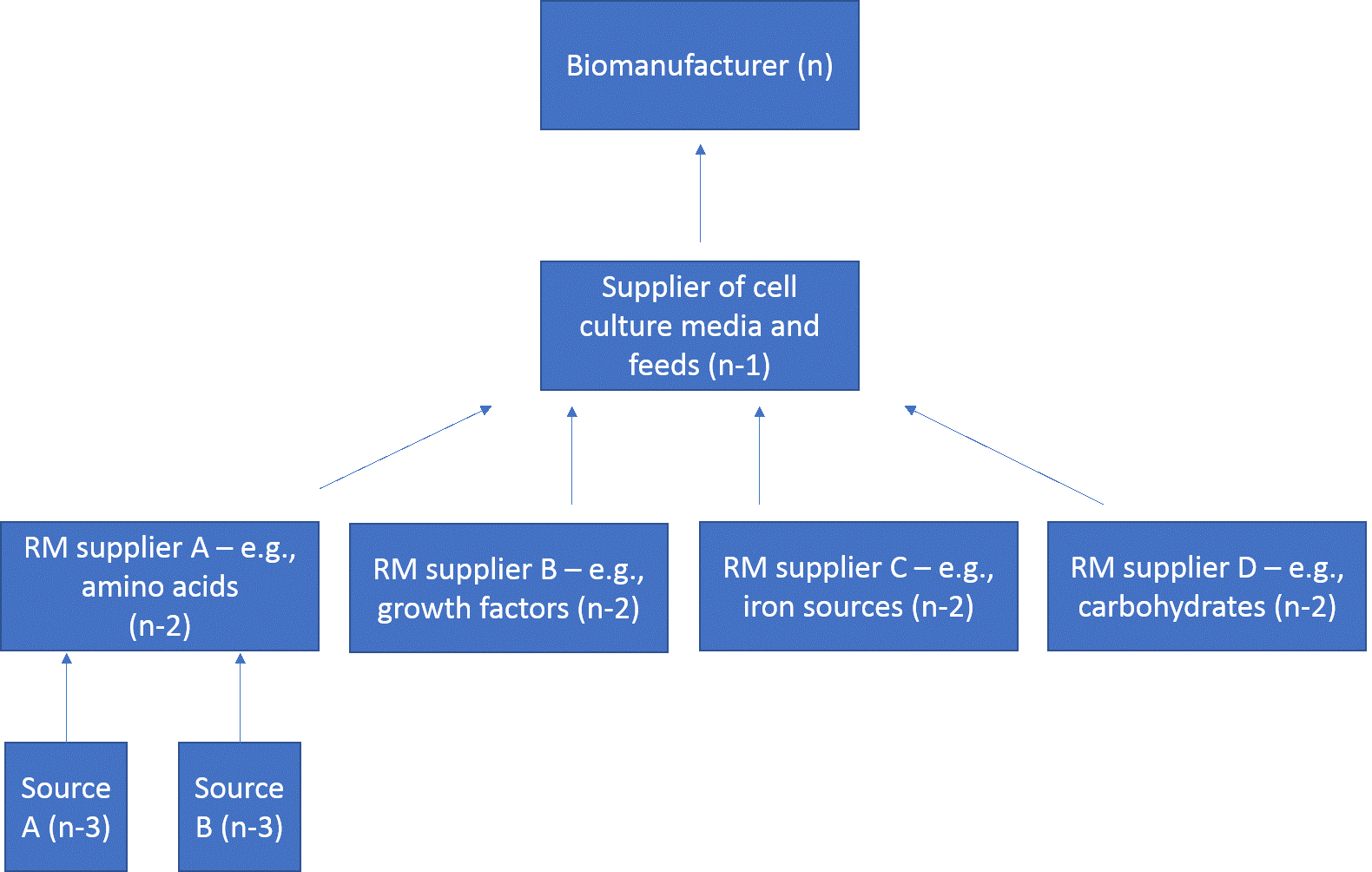
Fig 1. A simplified supply chain model for bioprocess material sourcing.
Raw material risk assessment at our company
We source thousands of raw materials for producing cell culture media and feeds. Because of the vast number of varying types of materials flowing through receiving inspection, a comprehensive characterization program is not realistic. All raw materials, including compendial grades, must meet baseline requirements and testing per specifications before they are accepted into inventory. However, because raw materials are prioritized based on risk to process and product, those materials identified as high risk are further characterized using a set of defined parameters.
We evaluate raw chemical components used in cell culture media and feeds based on rankings in the following risk categories:
- Traceability and origin country’s regulatory environment. The country and supplier of origin of each raw material are important factors to consider because some countries might not have sufficient regulatory requirements, and suppliers could lack adequate quality management and/or traceability for biomanufacturers’ needs. Another factor is whether sourcing is direct or indirect (i.e., through a distributor). Suppliers with rigorous quality management systems and traceability programs are preferred; materials from these suppliers generally receive lower risk scores.
- Manufacturing technique. Components for cell culture media and feeds can be endogenous, synthesized, mined, or recycled. Endogenous and synthesized materials are classified as animal-derived or animal-derived component-free (ADCF). ADCF components can be produced by cell culture or fermentation or they can be derived from plants. Animal-derived components are typically assigned higher risk scores than materials not derived from animals.
- Assurance of supply. Security of supply is important to ensure biomanufacturers have sufficient materials to maintain production. When evaluating suppliers, several points are considered. Among these are the number of sources they use (n-3), their willingness to provide us with supply contracts, and the robustness of their change control process. Suppliers with strong assurance of supply programs are given preference and typically receive lower risk scores.
- Component criticality. Factors that determine criticality include a component’s predominance in cell culture media and the likelihood that variation will cause failure. Characterization is key to identifying and measuring impurities in these components. In general, the more critical a component is, the higher the assigned risk score.
- Annual acceptance rate and usage. Considerations include history of acceptance and failures, as well as number of lots we use per year. In general, the risk score increases as usage increases. Characterization is essential to ensure low lot-to-lot variability in impurity levels of materials with high annual usage.
- Compendial test availability. Components accompanied by data for compendial specifications (e.g., USP, NF, EP, ACC, JP) typically receive lower risk scores. Although compendial test methods are outdated, this data does provide some indication of component quality.
Risks assessed for raw materials, specific amino acids, iron carriers, and excipients were rated as higher than average risk and under greater scrutiny. In our risk assessment scheme, raw materials with higher risk scores are characterized further. We continually work to mitigate and reduce risks of high-scoring materials, with the goal of reducing the number of materials considered high risk.
Risk mitigation
Risk mitigation methods vary depending on the component type. Animal-derived components are under increased scrutiny due to high variability and risk of viral or other adventitious agents. Careful consideration of supplier and source is essential to minimizing impacts of variability. Another option is to mitigate this risk by using animal-derived component-free media. For example, Cytiva's chemically defined media do not contain animal-derived components.
Oftentimes, risks can be mitigated by effective supplier management. One potential action to mitigate risk is to identify suppliers (n-2) with the cleanest, highest-grade materials. However, as mentioned previously, lot selection is not an ideal solution and should be used only when a more strategic and permanent solution has not been identified. An example of a strategic and potentially permanent solution that we continually seek is to collaborate with suppliers by sharing characterization information and working with them to identify possible solutions based on the lot characterization profiles. For instance, an n-2 supplier might source material from three different n-3 suppliers. Using the characterization profiles, it is possible the n-2 supplier can identify one or two of the n-3 suppliers that provide a lower impurity profile. Another potential avenue of collaboration to improve quality and consistency is the identification of process improvements made possible by sharing characterization data.
Purity and impurity analysis of an iron carrier
Characterization of iron carriers is important for several reasons. Iron sources, such as ferrous and ferric salts, are necessary components of chemically defined media. Variations in iron content (purity) can impact cell functions that require iron. Further, iron carriers are contaminated with other metals (impurities). Some of these metals can impact cell culture processes or product quality at levels of less than 10 parts per billion (ppb).
For the characterization study shown in Figure 2, an iron carrier was sourced from seven suppliers. Multiple lots were sourced from some suppliers, for a total of 14 lots. Samples of equivalent masses were prepared for elemental analysis. Seventy elements, including iron, were analyzed using inductively coupled plasma-mass spectrometry (ICP-MS). Results for 16 elements are provided in the figure.
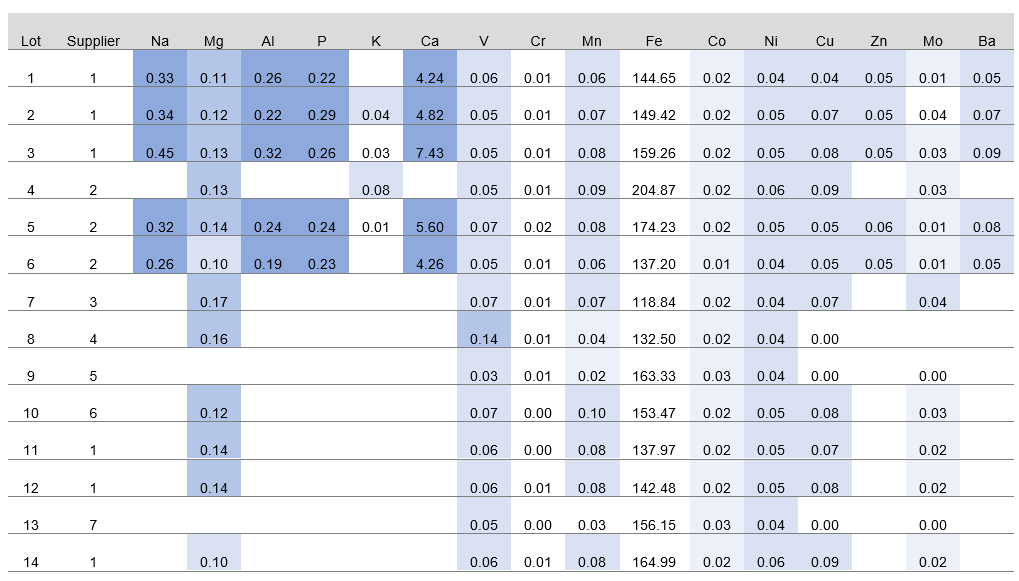
Fig 2. Elemental analysis of 14 lots sourced from 7 suppliers of an iron-containing compound commonly used in cell culture. Values are in parts per million (ppm).
Results showed substantial lot-to-lot variation in purity and impurity levels. Iron content averaged 152.8 ppm, ranging from 118.84 to 204.90 ppm. Major metal impurities were calcium (very high level of more than 1 ppm) and sodium, aluminum, and phosphorus (all three at high levels of more than 0.2 ppm). Calcium levels ranged from less than the limit of detection (LOD) to 7.43 ppm. The lowest levels for sodium, aluminum, and phosphorus were less than LOD; highest levels were 0.45, 0.32, and 0.29 ppm, respectively.
Summary
Today’s cell culture processes, using chemically defined media and highly controlled processes, reveal a growing impact from raw materials. Variation in raw materials can be significant, potentially causing process and product quality variation. A sound, risk-based approach for handling raw materials is increasingly important to control variability, according to the FDA and other regulatory guidelines. Ideally, such risks will be mitigated before affected raw materials are used in a biomanufacturing process. An organized effort across the biopharmaceutical industry, including drug substance manufacturers and suppliers, is needed to address and meet industry needs regarding raw material quality and consistency.
References
- International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. Q8(R2) Pharmaceutical Development (2009). https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf Accessed 24 August 2019.
- Food and Drug Administration, Guidance for industry, process validation: general principles and practices (CDER, Rockville, Md., 2011, p. 4). https://www.fda.gov/media/71021/download Accessed 24 August 2019.
- Understanding and modeling is a core component of modern drug development. By BioPharm International Editors. BioPharm International26 (2013). http://www.biopharminternational.com/understanding-and-modeling-product-and-process-variation. Accessed 24 August 2019.
- Hossler P. and Khattak S. F. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology19, 936–949 (2009). https://www.ncbi.nlm.nih.gov/pubmed/19494347