Many studies are evaluating natural killer (NK) cells as potential cancer treatments. However, it’s challenging to obtain primary human NK cells at clinically relevant scale. We generated 1 × 109 cells in a workflow amenable to a current good manufacturing practices (cGMP) environment. Throughout culture, the NK cells maintained > 97% purity, and after xeno-free expansion cells were shown to be activated. Here, we demonstrate the feasibility of xeno-free NK cell expansion and closure of key unit operations.
What is NK cell therapy?
Natural killer cells, discovered in the early 1970’s, represent only 5% to 20% of peripheral blood mononuclear cells (PBMCs; 1). Natural killer cells are phenotypically defined through the absence of CD3 expression and presence of CD56 (2, 3). Part of the innate immune system, NK cells efficiently kill a variety of malignant cells both in vitro and in vivo without prior sensitization (4–6). Malignant cell recognition is controlled by a complex balance of inhibitory and activating receptors on an NK cell’s surface. Natural killer cells have been receiving increasing attention (7, 8) as a cancer therapy, because they contribute to graft versus tumor (GvT) response but not to graft versus host disease (GvHD).
Approximately 5 × 106 to 1.2 × 107 NK cells/kg are needed to dose a patient suffering from a cancer such as multiple myeloma (9). One readily available source of NK cells is PBMCs collected from apheresis units. However, NK cells represent a small fraction of PBMCs in donors not treated with a mobilization agent. It’s challenging to obtain sufficient numbers to meet clinical requirements, especially when multiple infusions are planned. Isolating NK cells from PBMCs has become a major bottleneck that limits their clinical application. Therefore, protocols are being developed to address the need for larger cell numbers to meet clinical requirements.
NK cell production workflow
Here we describe a workflow that produces primary human NK cells at clinically relevant scale (Fig 1). CD3-/CD56+ cells were obtained using a functionally closed, semi-automated isolation process. Sepax™ C-Pro cell processing system with the BeadWash C-Pro application software was used in conjunction with a CliniMACS Plus system. Natural killer cells were then cultured without feeder cells under xeno-free conditions. After cells reached the desired volume and viable cell concentration, they were transferred into Xuri™ cell expansion system W25. At the end of culture, cells were harvested, volume-reduced, washed, and formulated into 100% CS10 medium using Sefia™ S-2000 cell processing system, which supports automated and functionally closed processing. Formulated cells were then cryopreserved using the liquid nitrogen-free VIA Freeze™ controlled-rate freezer. Cultures were periodically tested for their characteristic phenotype and functional killing efficacy.
Overall, the feasibility of xeno-free expansion of primary human NK cells was demonstrated. Natural killer cells were 90% pure after the isolation step. Throughout the cell expansion step, NK cells maintained > 97% purity, and after expansion cells were shown to be activated. Feasibility of using functionally closed systems for harvest and cryopreservation was also shown. To further enhance the good manufacturing practices (GMP) readiness of the workflow, a separate experiment evaluated the possibility of using growth factors without MicroBeads for NK cell activation.
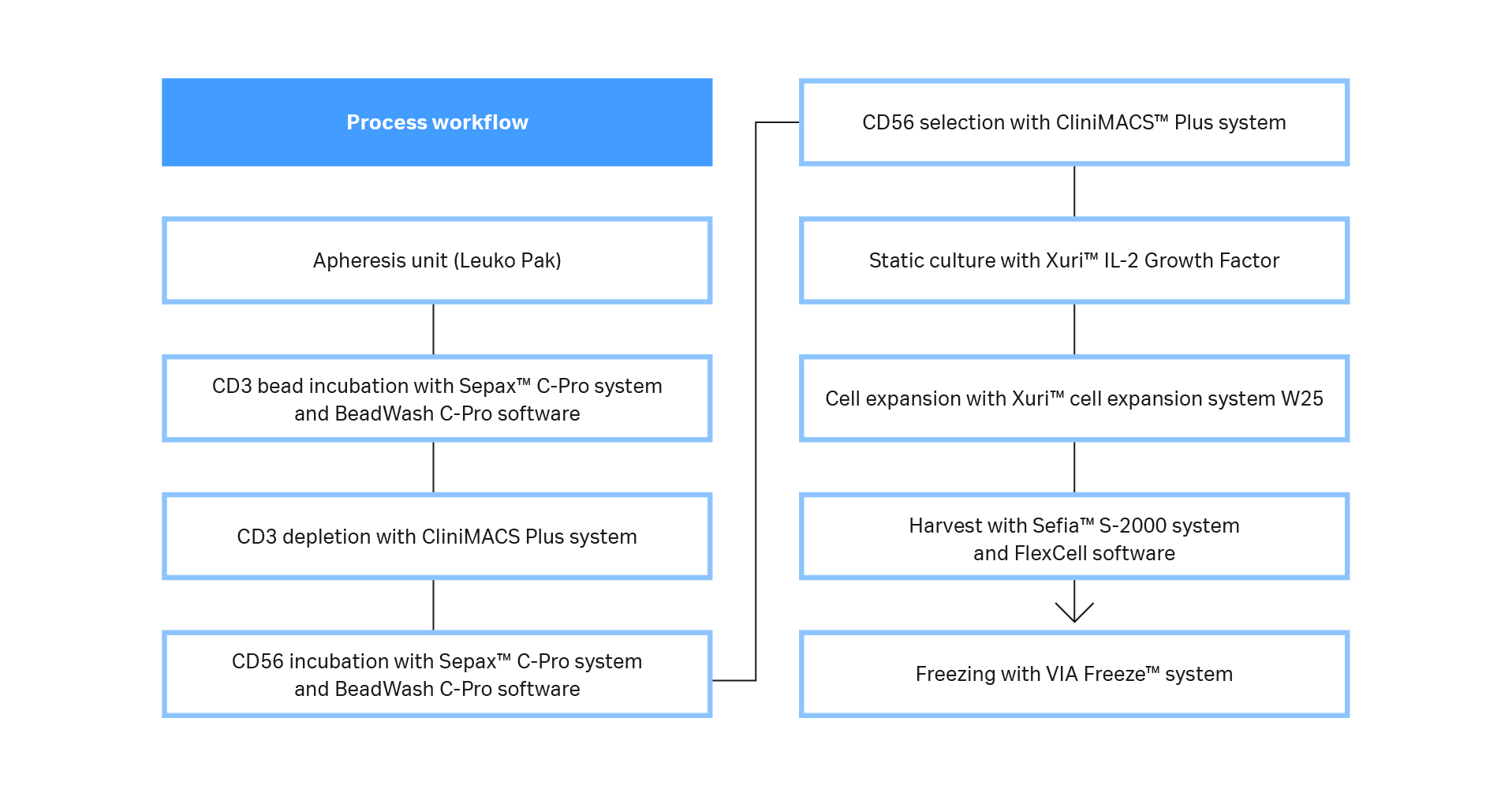
Fig 1. Process workflow. A diagram of the steps and systems used to prepare clinically relevant doses of NK cells from fresh leukapheresis units. Most steps were functionally closed and automated, and the process is compatible with GMP guidelines.
Materials and methods are available in the Related information section at the bottom of page.
Results and discussion – large‑scale NK cell production
As expected, expansion potential varied between donors to a substantial degree. Populations of pure NK cells were obtained by magnetic separation using the CliniMACS system. Natural killer cell purity increased from between 4% and 11% in initial populations, to above 90%, regardless of different starting populations of cells (Fig 2A, B). A high-purity NK population is particularly important for allogeneic administration, because it reduces the risk and severity of GvHD.
Currently, CliniMACS requires open incubation of MicroBeads and cells in a tissue culture hood. Here, we performed a functionally closed and automated incubation using the Sepax C-Pro cell processing system with BeadWash C-Pro software. The recovery from the initial CD3 MicroBead incubation ranged from 57% to 73.5% (average 65%; Fig 2C). These recoveries are consistent with expectations after platelet removal of leukapheresis units. Recovery after CD56 MicroBead incubation ranged from 98% to 103% (average 100%, Fig 2C).
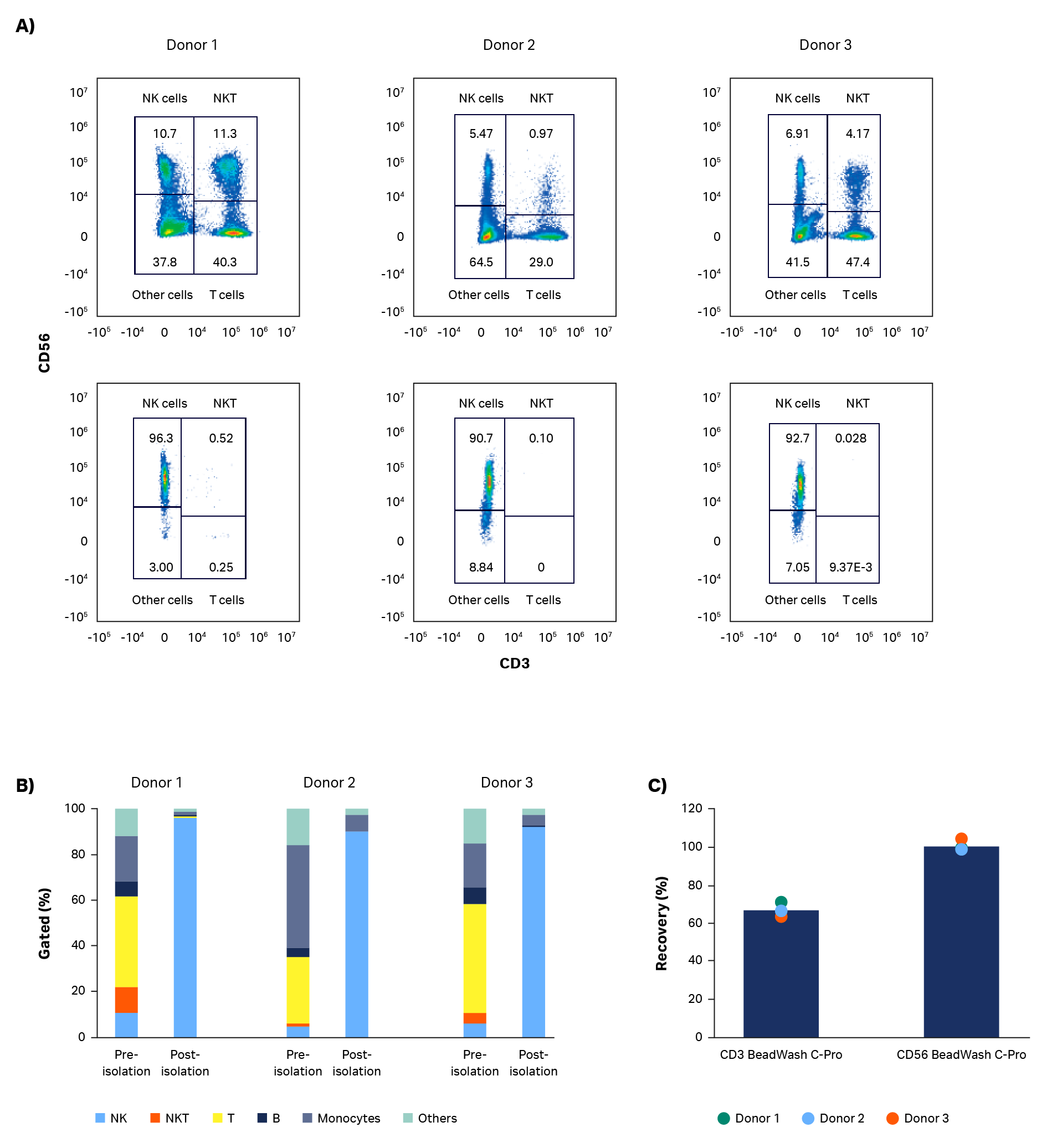
Fig 2. Purity and recovery of NK cells after isolation. Phenotypic characterization of apheresis products before and after NK cell isolation procedure. (A) FlowJo representation of gated cells from each donor on day 0. (B) Measurements of isolated cells on day 0 using flow cytometry from donors 1, 2, and 3 showing NK cells (CD56+/CD3-), T cells (CD56-/CD3+), NK-like T cells (NKT, CD56+/CD3+), and other cells such as monocytes (CD14+) and B cells (CD19+). (C) Viable cell recovery after CD3 or CD56 MicroBead incubation with BeadWash C-Pro application on Sepax™ C-Pro system. Bars represent the average of biological triplicates with individual donor values shown by dots.
After isolation, cells were expanded in a xeno-free process. Cells were cultured in a commercially available xeno-free medium containing Xuri™ IL-2 and NK Activation/Expansion Kit beads. The cells expanded steadily during static culture, with continued growth after transfer to the Xuri cell expansion system W25. Donor 1 cells grew particularly well in the Xuri Cellbag™ bioreactor (almost 10-fold from static culture). Donor 2 and 3 cells approximately tripled and doubled, respectively. The cumulative cell growth reached between 2.6 × 109 and 7.4 × 109 viable total cells by day 20 (Fig 3A). Natural killer cell purity was maintained above 97% throughout the expansion step (Fig 3C), resulting in 2.5 × 109 to 7.2 × 109 live NK cells on day 20 (Fig 3B).
Phenotypic analysis was performed to examine expression of some key NK receptors and activation markers (Fig 3C). NKg2D is constitutively expressed in natural killer cells; expression on NK cells was measured at > 93% throughout the entire step. NKp44 (NCR2) is a natural cytotoxicity receptor that is upregulated on activated natural killer cells. CD69 is an activation marker that is one of the earliest inducible cell surface glycoproteins on T cells and NK cells. In these culture steps, NKp44 and CD69 were expressed in less than 5% of NK cells at culture start. After natural killer cell activation and until the end of culture, NKp44 and CD69 were expressed in over 90% of NK cells.
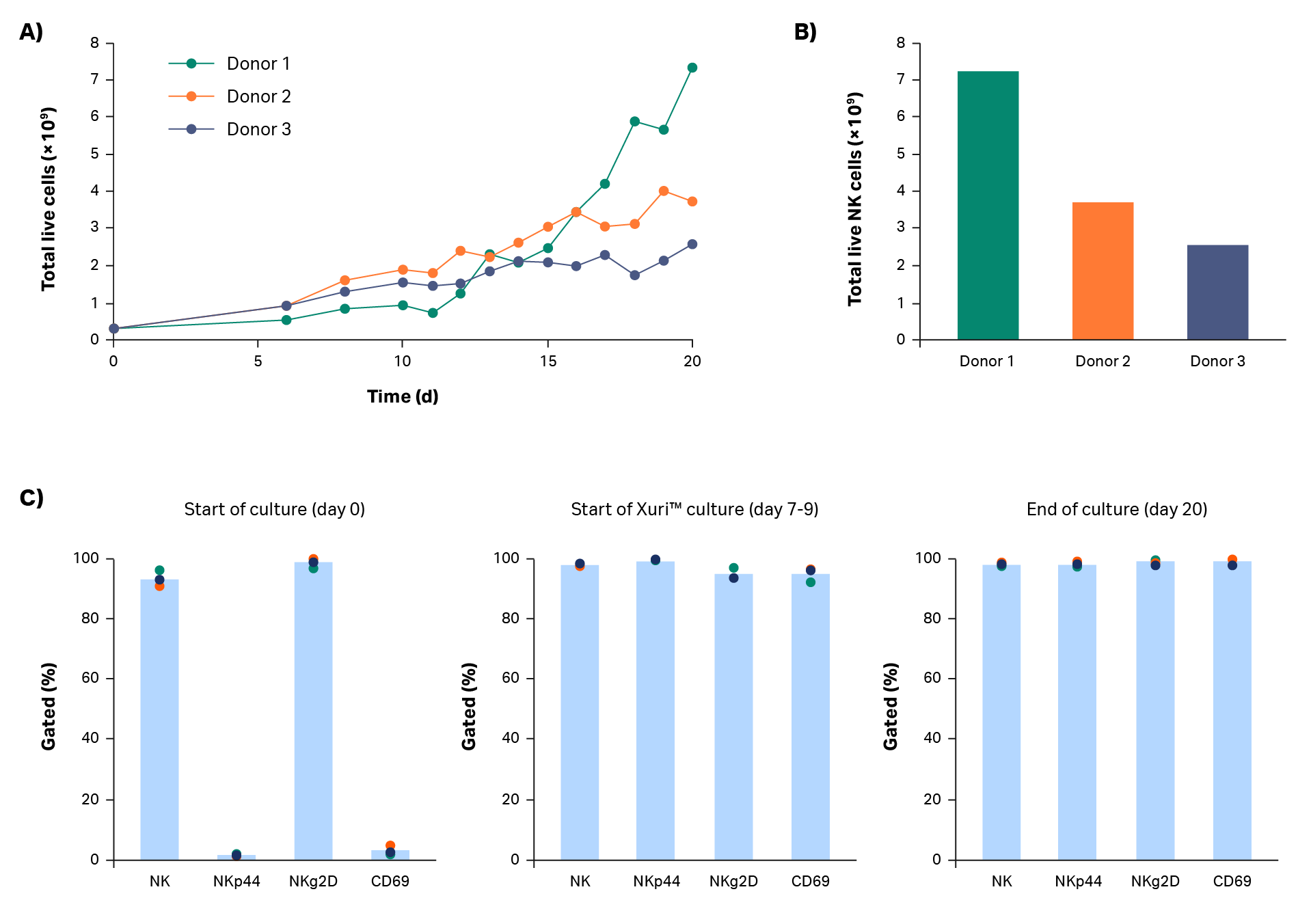
Fig 3. Culture growth and phenotype. Cumulative growth of (A) total live cells and (B) NK cells on day 20 for donors 1, 2, and 3. (C) Measurement of NK cell (CD3-/CD56+) subpopulations NKp44+, NKg2D+, and CD69+ cells by flow cytometry before, during, and after cell expansion. Bars represent the average of biological triplicates with individual donor values shown by dots.
After expansion, cells must be washed and formulated in a buffer suitable for immediate infusion into the patient or, alternatively, into a cryoprotectant. Formulation with a cryoprotectant enables cryopreservation and subsequent transportation to the clinical site.
In this workflow, expanded natural killer cells were harvested using the Sefia S-2000 system prior to cryopreservation. This functionally closed, automated, centrifugation-based system can harvest, wash, and resuspend cells into cryoprotectant. After volume reduction and wash with the Sefia S-2000 system, total live cell recovery was 97%, 91%, and 80% for donors 1, 2, and 3, respectively (Fig 4A). An average decrease in viability of 8.5% was observed after harvest (p = 0.0034 by 2-sample t-test, n = 3, Fig 4A). Optimization of parameters such as the centrifugation speed and time could mitigate this decrease.
Phenotypic comparison of cells pre- and post-harvesting in the Sefia S-2000 system is shown in Figure 4B. No statistically significant differences were found in natural killer cell purity or expression of NK markers NKg2D/NKp44 and cell activation marker CD69 (p > 0.05 by 2-sample t-test, n = 3).
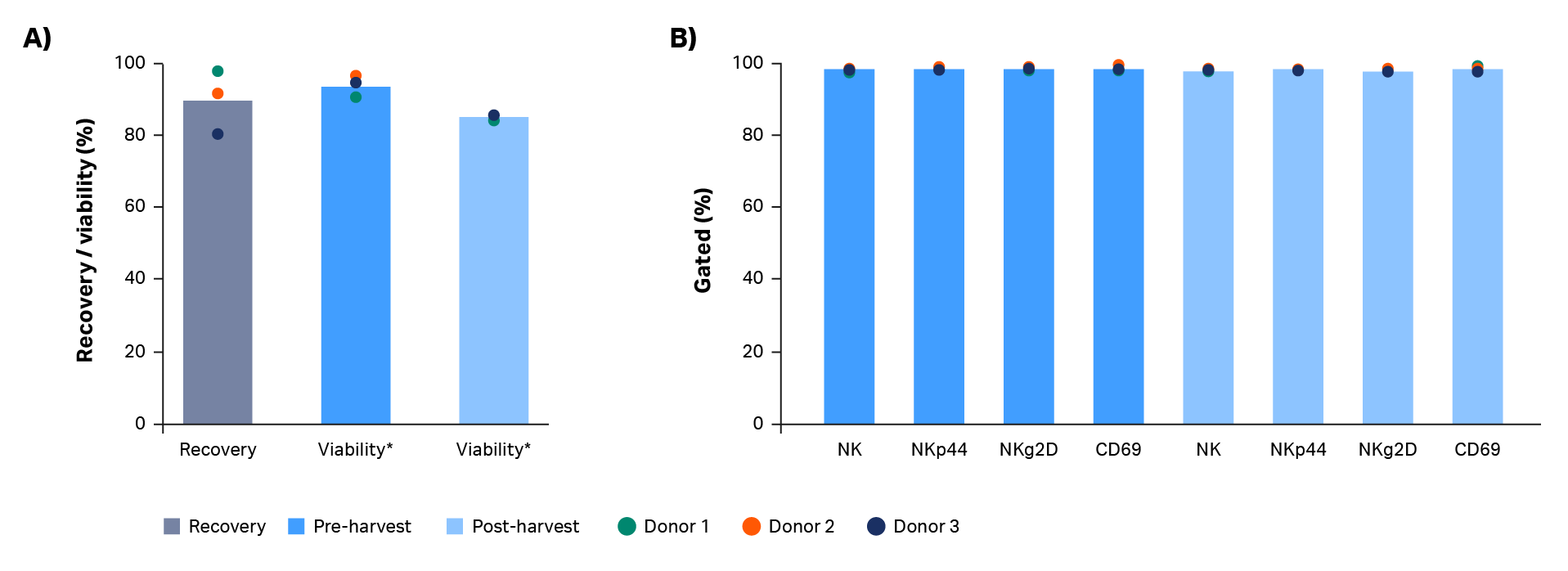
Fig 4. Recovery and phenotype of NK cells before and after Sefia™ S-2000 system harvest. Characterization of NK cells before and after Sefia S-2000 system harvest. (A) Recovery and viability of live cells from Donor 1, 2, and 3. (B) Percentage of NK cells (CD3-/CD56+) and their subpopulations (NKp44+, NKg2D+, CD69+) before vs after harvest. Bars represent the average of biological triplicates with individual donor values shown by dots.
* p ≤ 0.05 by 2-sample t-test. n = 3.
NK cytotoxicity against K562 cells was assessed at the start and end of the expansion step using a flow cytometry-based approach. After 5 h of co-culture, target cell death was determined. Results are presented in Figure 5 (representative scatter plots and gating analysis for one donor shown in Fig 5A–C, results presented in Fig 5D). Day 1 resting NK cells have limited ability to kill target cells even at a 5:1 effector:target (E:T) ratio (< 15% cell death, Fig 5D). However, by day 21, target cell killing was > 70% at the 5:1 E:T ratio. These results indicate that functionally activated NK cells were present even after expansion. A comparison of NK cell killing ability was also carried out pre- and post-harvest in the Sefia S-2000 system (Fig 5D). As with phenotype, there were no statistically significant differences in killing ability after harvest (p > 0.05 by 2-sample t-test; n = 3).
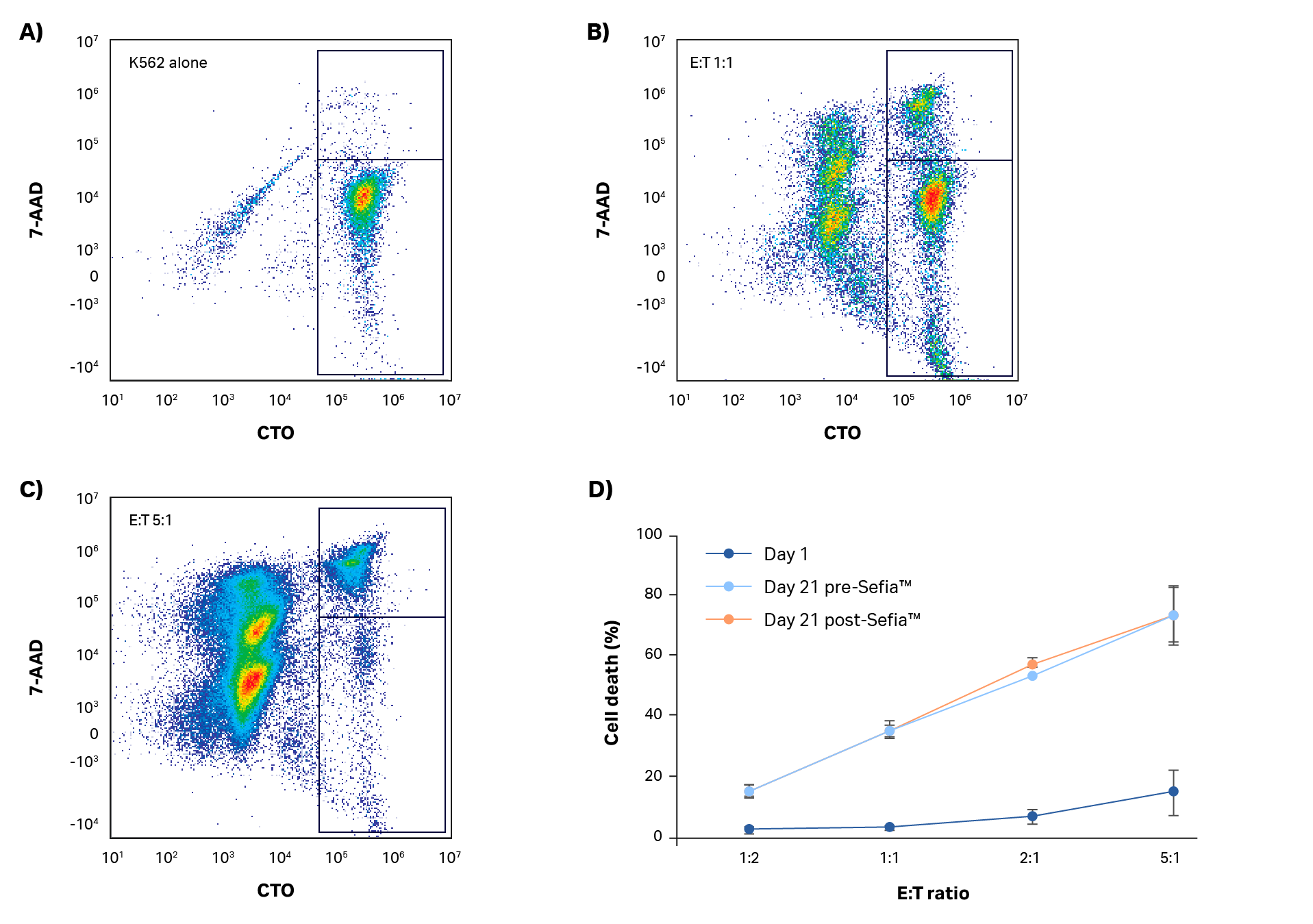
Fig 5. Killing assay. (A–C) Example gating analysis and (D) quantification of NK cell cytotoxic activity in response to K562 co-culture at different effector:target (E:T) ratios after 5 h incubation. Day 1 and day 21 cultures before and after harvest with Sefia S-2000 system are shown. No statistically significant differences in NK potency were measured after Sefia™ S-2000 system processing. Error bars represent standard deviation. P > 0.05 by 2-sample t-test. N = 3.
After harvesting with Sefia S-2000 system, cells were cryopreserved in CS10 medium using the VIA Freeze liquid nitrogen-free freezer, an alternative to static containers and liquid nitrogen-based freezing methods.
Thawing of cryovials resulted in recoveries and viabilities > 70% on average (Fig 6A). Cryopreservation and thaw processes had no effect on NK cell phenotype, which was maintained from the end of culture (Fig 6B). To improve recoveries, further optimization could be performed on formulation, cooling rates, and thawing steps.
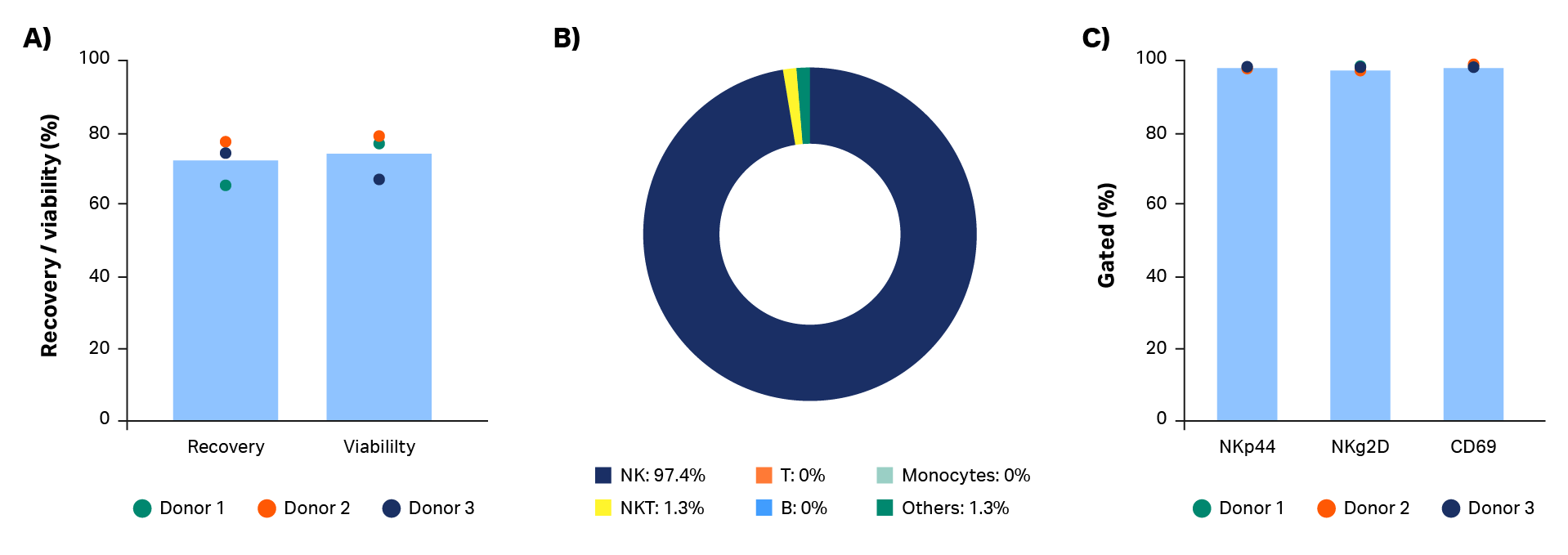
Fig 6. Cryopreservation and thawing. A) Post-thaw recovery and viability of NK cells cryopreserved in cryovials with 100% CS10. (B) Thawed NK cells maintain purity and (C) an activated NK cell phenotype at the end of culture. Bars represent the average of biological triplicates with individual donor values shown by dots. n = 3.
Results and discussion – comparison of activation methods
Several methods are available for expanding natural killer cells with antigen-presenting cells. The use of feeder cells has been shown to provide high fold-expansion. However, the use of such feeder systems is unlikely to be amenable to scale-up. Also, feeder cells might pose risks to the patient if not properly removed from the co-culture. Another option is to use magnetic particles, but current methods do not comply with GMP manufacturing requirements. Alternatively, high-quality cytokines can be used to activate and expand cells. Xuri IL-2, Xuri IL-15, and Xuri IL-21 growth factors follow USP <1043> for cell therapy ancillary material.
A combination of cytokines (Xuri IL-2, Xuri IL-15, and Xuri IL-21 growth factors at concentrations of 350 IU/mL, 66.7 IU/mL, and 0.341 IU/mL, respectively) were tested for their ability to activate and expand natural killer cells compared to NK Cell Activation/Expansion Kit beads and IL-2 (Fig 7). Manual isolation with the QuadroMACS system resulted in high purity (> 90%) NK cell cultures in all four donors (Fig 7C). Despite donor-to-donor variability noted in average daily fold-expansion, the use of Xuri growth factors resulted in expanded NK cells (Fig 7A). However, use of IL-2 in combination with the NK Cell Activation/Expansion Kit generally had higher daily fold-expansions compared with cultures using cytokines alone. The differences in results were not statistically significant (p ≥ 0.05 by 2-sample t-test, n = 4).
The activation and expansion of natural killer cells with cytokines rather than activation beads resulted in comparable viability (Fig 7B, p ≥ 0.05 by 2-sample t-test, n = 4) and purity (Fig 7C; p ≥ 0.05 by 2-sample t-test, n = 4). Further optimization of cytokine concentrations, timing of addition, as well as other cytokine combinations could lead to more robust NK cell activation and expansion.
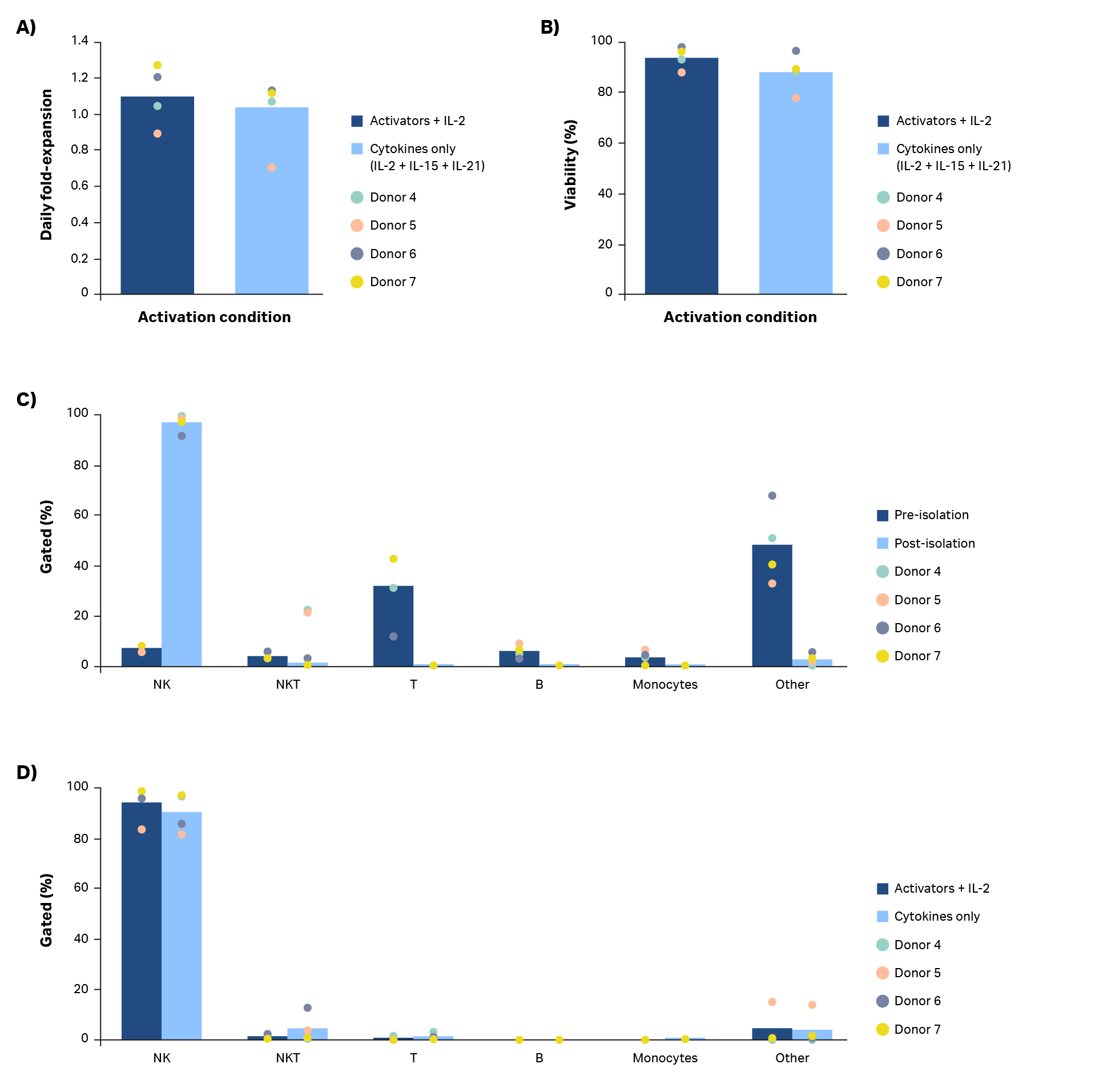
Fig 7. Comparison of activation methods. (A) Average daily fold-expansion over culture of NK cells activated using a combination of Xuri IL-2, Xuri IL-15, and Xuri IL-21 growth factors compared with cells activated using an NK Activation/ Expansion Kit supplemented with cells activated using an NK Activation/ Expansion Kit supplemented with IL-2 and (B) corresponding % viability on day 20. (C) Phenotypic characterization of cultures on day 0 and (D) day 20 from flow cytometry. Bars represent the average of biological replicates with individual donor values shown by dots.
Conclusions
We presented a workflow for xeno-free natural killer cell expansion from leukapheresis units. The process incorporates several automated and functionally closed unit operations (Fig 1). Despite substantial heterogeneity and donor-to-donor variability of the input cell population, magnetic isolation followed by activation and expansion to clinically relevant doses (> 109 cells per donor) resulted in a highly pure (> 97% CD3-/CD56+) NK cell product. Phenotypically and functionally, the final cellular product demonstrated characteristics of an activated NK population with robust cytotoxic activity against a tumor cell line.
Overall, the methodology presented here is compatible with industrial workflows for natural killer cell manufacturing. This white paper shows the feasibility of xeno-free NK cell expansion and closure of key unit operations.
Future work could focus on fine-tuning each step to generate a robust, scalable process. For example, cell recovery and viability could be enhanced by optimizing the activation strategy, cryopreservation parameters, and other culture parameters. Also, improvements to reagent packaging, as well as static culture in gas-permeable bags or closed flasks, might enable complete closure of the workflow.
The small-scale experiment from buffy coats shows the possibility of cytokine-mediated activation. Optimization of this step could lead to an activation process amenable to cGMP manufacturing environments.
Acknowledgements
All work was performed in collaboration with CCRM through funding from FedDev Ontario and Cytiva at the Centre for Advanced Therapeutic Cell Technologies (CATCT), Toronto, Ontario, Canada. We are grateful to Canadian Blood Services and donors for providing research samples for completion of this project. The reporting and interpretation of the research findings are the responsibility of the author(s). The views expressed herein do not necessarily represent the views of Canadian Blood Services.
References
- Herberman, R.B. et al. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. characterization of effector cells. Int J Cancer16:230–239 (1975).
- Cooper, M.A. et al. The biology of human natural killer‑cell subsets. Trends Immunol. 22:633–640 (2001).
- Barao, I. and Ascensao, J. L. Human natural killer cells. Arch. Immunol. Ther. Exp. Warsz. 46:213–229 (1998).
- Lee, S. H. et al. Keeping NK cells in highly regulated antiviral warfare. Trends Immunol. 28:252–259 (2007).
- Marcus, A. et al. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 122:91–128 (2014).
- Shook, D. R. and Campana, D. Natural killer cell engineering for cellular therapy of cancer. Tissue Antigens 78:409–415 (2011).
- Pittari, G. et al. Revving up natural killer cells and cytokine‑induced killer cells against hematological malignancies. Front. Immunol. 6:230 (2015).
- Rezvani, K. and Rouce, R. H. The application of natural killer cell immunotherapy for the treatment of cancer. Front. Immunol. 6:578 (2015).
- Cheng, M. et al. NK cell‑based immunotherapy for malignant diseases. Cell Mol. Immunol. 10:230–252 (2013).
CliniMACS Plus system and QuadroMACS system are trademarks of Miltenyi Biotec GmbH. FlowJo is a trademark of FlowJo LLC. Any other third-party trademarks are property of their respective owners.
Ordering information
| Product | Product code |
| HyClone Penicillin‑Streptomycin 100X solution | SV30010 |
| HyClone Dulbecco’s Phosphate Buffered Saline (DPBS), 10× | SH30378 |
| Xuri IL‑2 Growth Factor | 29062789 |
| Xuri IL‑15 Growth Factor | 29112116 |
| Xuri IL‑21 Growth Factor | 29112119 |
| Sepax C‑Pro Cell Processing Instrument | 29264741 |
| BeadWash C‑Pro Protocol Software | 29734656 |
| Xuri Cell Expansion System Cellbag Perfusion, 2 L | 29108442 |
| Xuri 1.2 µm Perfusion Filter Cellbags | 29279170 |
| Xuri Cell Expansion System W25 | 29064568 |
| Sefia S‑2000 Cell Processing Instrument | 29285527 |
| FlexCell Sefia Protocol Software | 16301 |
| VIA Freeze Duo Controlled‑Rate Freezer | VFD_30006 |
Materials and methods – large-scale NK cell production
NK cell isolation
NK cell isolation involved a CD3 depletion step followed by CD56 selection. Three full Leuko Pak (STEMCELL Technologies) were each incubated with CliniMACS CD3 MicroBeads (Miltenyi Biotec) in the Sepax C‑Pro Cell Processing System (Cytiva) using the BeadWash C‑Pro protocol software (Table 1). The output bag from the BeadWash C‑Pro protocol was connected via a spike port to the CliniMACS LD Depletion Tubing Set (Miltenyi Biotec) and run on the CliniMACS Plus System using the Depletion Protocol 3.1 according to the manufacturer’s instructions.
The CD3‑ fraction was then incubated with CliniMACS CD56 MicroBeads (Miltenyi Biotec) in Sepax C‑Pro using the BeadWash C‑Pro protocol software (Table 1). The output bags from the BeadWash C‑Pro protocol were welded onto a CliniMACS Tubing Set LS (Miltenyi Biotec) and run on the CliniMACS Plus Sytem using the CliniMACS Protocol 1.1 Enrichment according to the manufacturer’s instructions.
Xeno-free static culture
NK culture medium was prepared by supplementing a commercially available xeno-free medium with HyClone Penicillin‑Streptomycin solution diluted to working concentration (1% P/S, Cytiva). Natural killer cells were activated using NK Activation/Expansion Kit (Miltenyi Biotec) according to the manufacturer’s protocol and supplemented with 350 IU/mL Xuri IL‑2 Growth Factor (Cytiva). Cells were inoculated into tissue culture‑treated multiwell plates or flasks and seeded at a density of 1 × 106 cells/ mL. Cells were cultured in an incubator (37°C, 5% CO2) for 6 d to activate before further manipulations were performed.
After 6 d cells were counted and diluted to 1.5 × 106 cells/mL. Cells were kept in static conditions until total culture volume reached 2 × 106 cells/mL in at least 500 mL volume, which took 8 to 10 d. Then, cells were transferred to a Xuri Cell Expansion System W25.
Xeno-free expansion in Xuri W25 expansion system
A 2 L Xuri Cellbag bioreactor (Cytiva) was preconditioned with 100 mL each of fresh and conditioned culture medium (total volume 200 mL). The medium was the same formulation used in static culture. To prepare the conditioned medium, 100 mL of the NK cell suspension from a flask culture was centrifuged (400 × g, 5 min). The supernatant was then transferred to a 600 mL transfer pack (Fresenius Kabi). The Cellbag bioreactor was equilibrated at 37°C for 2 h before inoculating NK cells at 2 × 106 cells/mL.
Sampling was done each day, and medium was added until the total volume reached 1 L. Then, perfusion was initiated based on viable cell densities (Table 2). These perfusion rates were meant
to maintain lactate levels below 25 mM. The Cellbag bioreactor was agitated at 6 rpm and an angle of 6° for the entire duration of culture in the Xuri W25 expansion system.
Harvest
Natural killer cells were harvested using the Sefia S‑2000 Cell Processing System (Cytiva) and FlexCell protocol software. Briefly, NK cells were reduced in volume from 1 L to 20 mL at a 75 mL/min flow rate. Then, two wash cycles were performed using Dulbecco’s phosphate‑buffered saline without calcium or magnesium (DPBS‑/‑, Cytiva), supplemented with 1 mM EDTA (Sigma) and 2% heat‑inactivated human serum AB (Gemini). Washes were performed at 400 × g for 5 min. Cells were then extracted in 100 mL of NK basal medium (same commercially available medium used for static culture, without supplement).
Table 1. BeadWash C‑Pro parameters
| Parameter | BeadWash C‑Pro CD3 depletion | BeadWash C‑Pro CD56 selection |
| Initial volume | 120 mL | 220 mL |
| Detect Initial volume | N | N |
| Dilution ratio | 0 | 0 |
| Dilution speed | 17 mL/min | 17 mL/min |
| Mix after dilution | 60 s | 60 s |
| Thaw bag validation | N | N |
| Hang bag validation | N | N |
| Input bag rinse | Y | Y |
| Input bag rinse volume | 100 mL | 100 mL |
| Pause input bag rinse | N | N |
| Optical cell detection | Y | Y |
| Pause/exchange wash bag | N | N |
| Intermediate volume | 30 mL | 30 mL |
| Pre-wash cycle | 1 | 0 |
| Pre-wash g force | 85 × g | 300 × g |
| Pre-wash time | 420 s | 300 s |
| Reagent volume | 30 mL | 30 mL |
| Incubation volume | 102.5 mL | 102.5 mL |
| Incubation time | 30 min | 30 min |
| Mix rest time | 6 s | 6 s |
| Pause/switch waste bag | Off | Off |
| Post wash cycle | 1 | 1 |
| Post incubation wash | N/A | N/A |
| Post wash cycle g force | 300 × g | 300 × g |
| Post wash cycle time | 900 s | 900 s |
| Final volume | 150 mL | 150 mL |
| Pause/switch resuspension | N | N |
| Manual extraction | N | N |
| Syringe for manual extraction | N | N |
Table 2. Perfusion rates for NK cells in Xuri W25 expansion system
| Viable cell density | Perfusion rate |
| 2.0 × 106 cells/mL | 0.5 L/d |
| 1.0 × 107 cells/mL | 0.75 L/d |
| 1.5 × 107 cells/mL | 1 L/d |
Potency assay
NK cell potency was measured using a killing assay against an erythroleukemia cell line, K562 (ATCC). K562 cells were cultured in K562 medium, which was prepared by supplementing RPMI‑1640 (Thermo Fisher Scientific) with 5% heat‑inactivated fetal bovine serum (Sigma), 1% HyClone SG‑200 (stable L‑Alanyl‑L‑Glutamine; Cytiva), and 1% P/S. K562 cells were stained with 2.5 μM CellTracker™ Orange (Thermo Fisher Scientific) in K562 medium for 30 to 60 min at 37°C. The stained cells were washed twice in K562 medium and added to 96‑well round‑bottom plates with NK cells at different effector:target (E:T) ratios. A control well of K562 cells without NK cells was used to assess background cell death. Cells were incubated at 37°C for 5 h prior to centrifugation at 400 × g for 5 min. Supernatants were removed for 7‑amino‑actinomycin D (7‑AAD) staining (0.1 μg/ well in DPBS‑/‑; BD Biosciences). Cells were incubated for 5 min at ambient temperature before running on a CytoFLEX™ Flow Cytometer (Beckman Coulter). The specific cell death percentage was calculated using the following formula:

Phenotypic analysis
Natural killer cell populations were analyzed by flow cytometry using standard procedures. In brief, 1 × 106 cells were washed and blocked with human FcR Blocking Reagent (Miltenyi Biotec) prior to staining with antibodies from BD Biosciences: CD45‑APC‑Cy™7 (560178), CD14‑FITC (555397), CD3‑PE (555333), CD19‑APC (555415), CD56‑BV421 (562751), NKp44‑BB515 (566025), CD69‑BV650 (563834), and NKg2D‑APC (562064). Stained cells were analyzed on the CytoFLEX system.
Cryopreservation
NK cells were centrifuged at 400 × g for 5 min and resuspended in cold (4°C) 100% CryoStor™ CS10 (BioLife Solutions) to achieve a density of 2.5 × 107 cells/mL. The suspension was transferred to cryovials in 1 mL aliquots and then placed into a cooled rack (4°C) within the VIA Freeze Duo liquid nitrogen‑free, controlled‑rate freezer (Cytiva). The VIA Freeze protocol was run to cool the cells at ‑1°C/min until the temperature reached ‑100°C. Then, the vials were transferred to a liquid nitrogen tank for long‑term storage in the vapor phase. Cell recovery, viability, and phenotype were assessed after vials were thawed. Cell recovery and viability were determined using a NucleoCounter™ NC‑200. Phenotype was determined using flow cytometry.
Materials and methods – comparison of activation methods
Buffy coats from four healthy donors (Donors 4, 5, 6, and 7) were purchased from Canadian Blood Services. Buffy coats were processed using Ficoll‑Paque PLUS (Cytiva) to isolate PBMCs according to the manufacturer’s protocol. NK cells were enriched from PBMCs using the QuadroMACS™ Separator (Miltenyi Biotec). Briefly, CD3 cells were depleted using CliniMACS CD3 MicroBeads and run through an LD Column (Miltenyi Biotec). The CD3‑ fraction was then incubated with CliniMACS CD56 MicroBeads (Miltenyi) and run through an LS Column (Miltenyi Biotec). The positive fractions contained the enriched NK cells.
The enriched natural killer cells were activated and expanded by using the NK Cell Activation/Expansion Kit (Miltenyi Biotec), a combination of growth factors, or both methods together. The growth factor combination contained Xuri IL‑2, Xuri IL‑15, and Xuri IL‑21 (all Cytiva) at concentrations of 350 IU/mL, 66.7 IU/mL, and 0.341 IU/mL, respectively. Cells were counted every 2 d after day 6 and diluted to 1 × 106 cells/mL.