We adapted adherent HEK293T/HEK293 cells to suspension culture in our HyClone™ transfection medium, HyCell™ Transfx-H. The experiments we describe reveal information on cell growth by population doubling time, morphology, and viability necessary for future work for scalable production using single-use bioreactors.
The development of a transient transfection protocol that we describe is generic and can be used for different adeno-associated virus serotypes. The strategy was to use a Design of Experiments (DoE) methodology to evaluate several different parameters that could affect productivity and were measured as viral particles/L (VP/L), viral genomes/L (VG/L), and percentage full capsids.
Introduction
Viral vectors are increasingly used for gene transfer to specific tissue or to induce cell type modifications. Several viruses have been investigated for their use in cell and gene therapy with recombinant adeno-associated virus (rAAV) as the most promising vector for gene therapies.
The cell and gene therapy industry strives to establish efficient large-scale production of viral vectors with current good manufacturing practice (cGMP). In the quest for the high amounts necessary for clinical use of rAAV, companies are moving away from production using adherent HEK293 cells to more scalable technologies using suspension cell culture. Furthermore, transient transfection protocols are also being optimized for larger scales and efficiency in suspension.
Here, we utilized a design of experiment (DoE) methodology to optimize rAAV production in a HEK293T suspension cell system. An efficient and scalable cell culture process for AAV production — initially using rAAV2 serotype as a model for rAAV — was verified also for rAVV5.
The experiments we describe are in two steps:
- First, we describe how to adapt cells to a new cell culture medium. Through these experiments, we evaluated cell growth by population doubling time (PDT), morphology, and viability. Furthermore, our DoE experiments allow us to evaluate the impact of multiple factors, such as plasmid concentration, cell densities and harvest time. These could be key factors for future work for scalable AAV production, from small-scale shake flasks up to 25 L single-use bioreactor systems.
- Second, we describe how to optimize the AAV transient transfection, which forms the basis for a scalable AAV production process in single-use bioreactors.
To produce rAAV-2, we use a triple plasmid transfection system that provides the genes essential for rAAV-2 production in HEK293 cells and variants. First is the pHelper plasmid, which carries the E2A, E4, and VA genes. The second is the replication and capsid (rep/cap) plasmid for specific serotypes. The third is the plasmid with the gene of interest (GOI – the GFP gene in this study) and the inverted terminal repeat sequences (ITRs), which are essential for maintaining the virus’s infectivity (Fig 1).
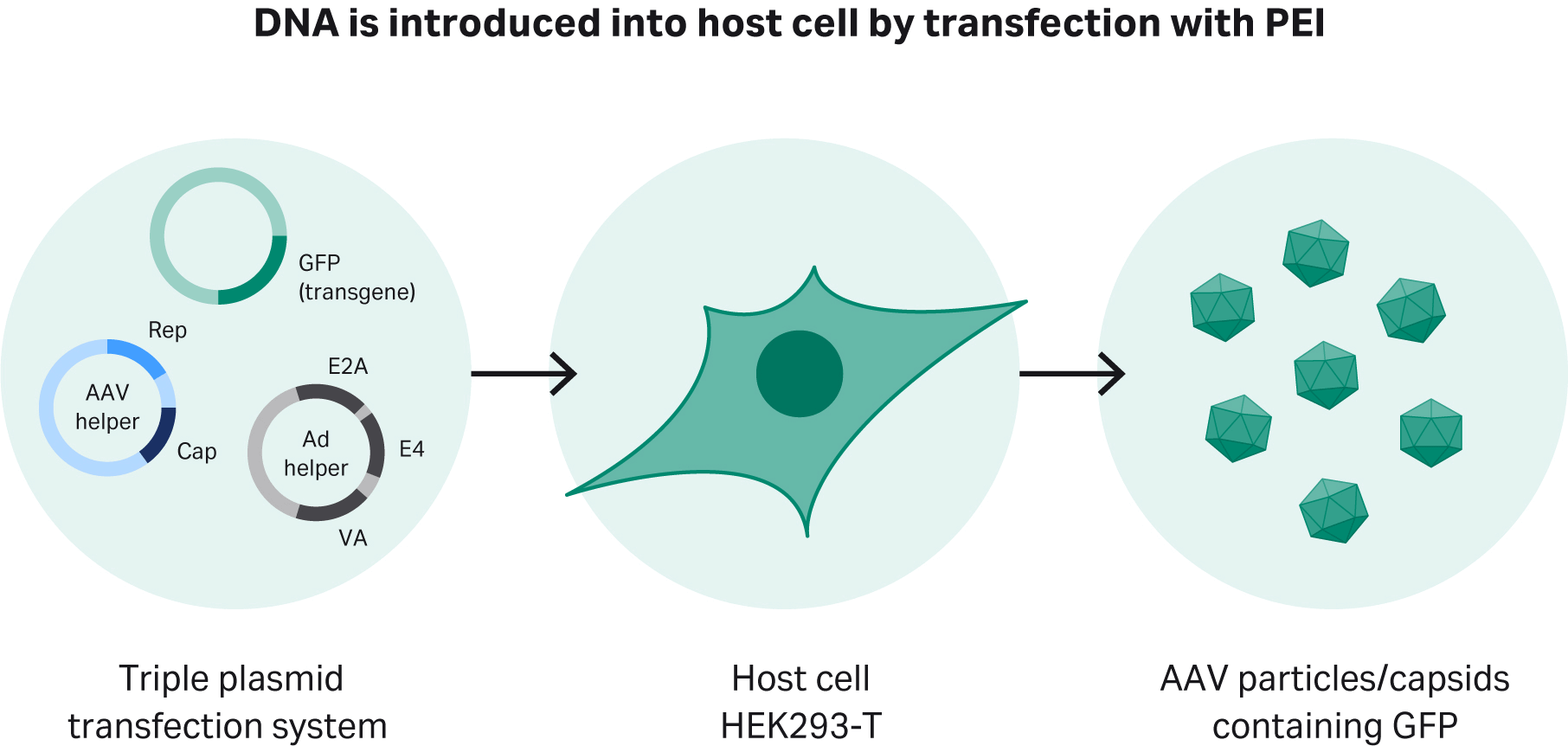
Fig 1. Transient transfection of adapted HEK293T cells using a triple plasmid transfection approach. Polyethyleneimine (PEI), forms a complex with DNA, which is then introduced into the host cells.
The upstream strategy we describe for AAV production in this application is summarized in Figure 2.

Fig 2. Our strategy is to develop a start-to-finish process for AAV production; we present a detailed account of the cell culture and transfection process development in this article.
Detailed Materials and methods are found at the end of this article.
Adaptation of HEK293T cells to a serum-free process for AAV production
We adapted adherent HEK293T cells to HyClone™ cell culture transfection medium, HyCell™ TransFx-H. The detailed protocol for the adaptation is described in Materials and methods. In parallel, HEK293 cells (without the SV40 large T antigen) were also successfully adapted to the same medium using the same strategy (data not shown).
Adaptation was performed by transferring cells grown in classical HyClone™ DMEM medium with high glucose supplemented and 10% HyClone™ Fetal Bovine Serum (FBS) directly into the new transfection medium. The medium was supplemented with 4 mM of HyClone™ L-glutamine and 0.1% Pluronic™ F-68 (Thermo Fisher Scientific™). The cell growth and morphology of the HEK293T suspension cells were evaluated microscopically over 10 passages in the respective serum-free media. To avoid aggregates and reduce shear stress, we used Pluronic™-F68 from the beginning of the adaptation and kept the cell density below 2.5 × 106 cells/mL.
Cells adapted to HyClone™ medium met our set success criteria of < 10% aggregation (small aggregates of < 10 cells) with robust cell growth. We created research and working cell banks (RCB and WCB, respectively). Cryopreserved cells from the HyCell™ TransFx-H medium were thawed from the cell banks created (see Materials and methods).
Thawing of cell bank and cell growth comparison
Cells were thawed from cell banks (see Materials and methods), seeded, and sub-cultivated twice a week.
Microscopic evaluation of cell morphology is shown in Figure 3. Most cells are viable and in single-cell suspension.

Fig 3. Single-cell suspension of HEK293T cells cultured in HyCell™ TransFx-H medium.
Population doubling time (PDT) was on average approximately 20 h, Figure 4. Good cell viability (> 97%) was maintained during 10 passages (Fig 4).
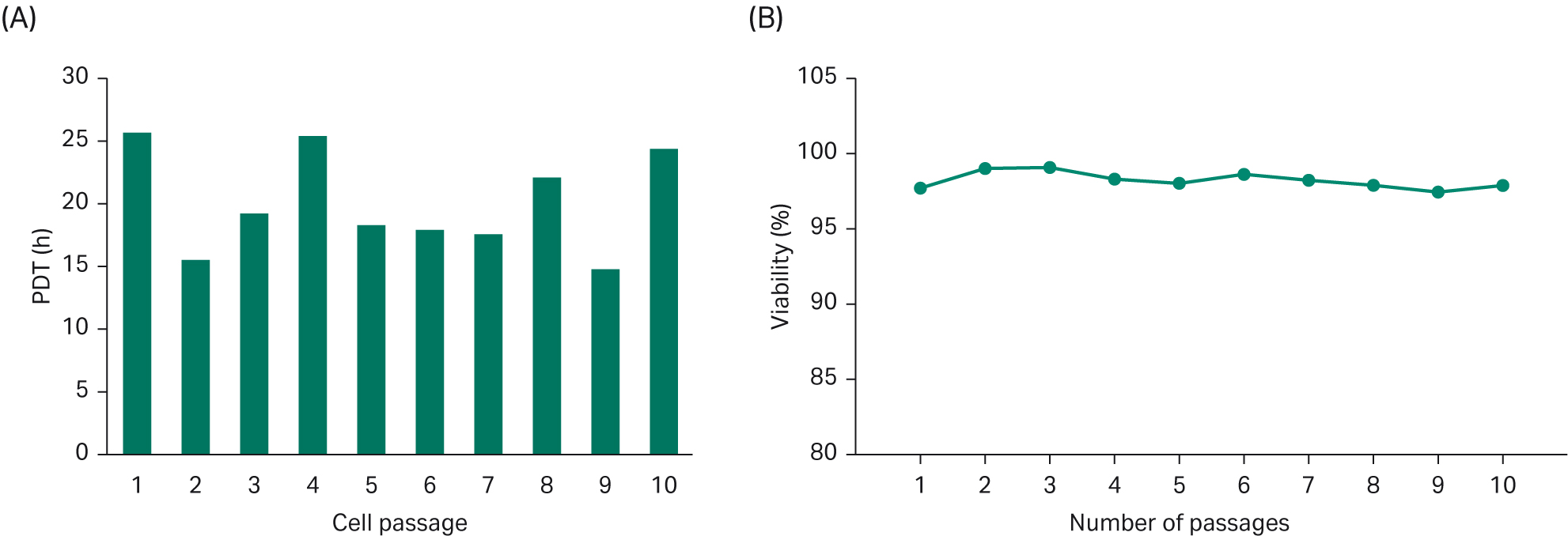
Fig 4. (A) Population doubling time for cells grown in HyCell™ TransFx-H media were studied over 10 passages. (B) Cell viability over 10 passages.
Plasmid constructs for AAV process development
Plasmid constructs containing the required genes and DNA sequences to produce AAV and used in our process development were originally obtained from Nordic Biosite.
Amplification of plasmids for AAV production is performed by transforming bacteria with plasmids. The plasmid DNA is subsequently replicated upon culturing the transformed bacteria. By using a plasmid purification kit, large amounts of plasmid DNA can be retrieved from the harvested material.
Design of Experiments (DoE); optimized rAAV production protocol using HEK293T suspension cells
A series of DoE studies were performed to evaluate the optimal conditions for the transfection. All in all, five studies were performed with four being applied to rAAV2. During our process development, the goal was to achieve the following criteria to meet the requirements for the downstream process: 1014 virus particles (VP)/L, 1013 viral genomes (VG)/L, and 10% or more full capsids in harvested material. DoE studies 1–4 showed that we could not achieve all these requirements for rAAV2, and consequently, the developed transfection protocol was tested for rAAV5. DoE5 was therefore designed as a verification study that confirmed the optimal DNA concentration and DNA-PEI complex incubation time for rAAV5 production.
DoE1
DoE1 investigated the plasmid DNA concentration, different cell densities, PEI/DNA ratio, transfection volume, and incubation time (Table 1). To verify the effect of different conditions, transfection efficiency was studied.
Table 1. Parameters used in the DoE1 setup
| Viable cell density (VCD) transfection (× 106/mL) |
DNA concentration (µg/mL) |
PEI/DNA (ratio) |
Transfection volume (% of total) |
Incubation time (min) |
|---|---|---|---|---|
| 1 | 1 | 6 | 10 | 45 |
| 0.75 | 0.75 | 4 | 6 | 30 |
| 0.5 | 0.5 | 2 | 2 | 15 |
In transient transfection, PEI condenses plasmid DNA into positively charged particles that interact with the anionic cell surface and the PEI/DNA complex enters the cells through endocytosis. The ratio of PEI/DNA have a key role in transfection efficiency. We evaluated all parameters in DoE 1 using MODDE™ Pro v12.0.1 software (Sartorius Stedim GmbH), which summarizes and evaluates all the data. The results allowed us to predict which parameters should be used to achieve high transfection efficiency (Fig 5).
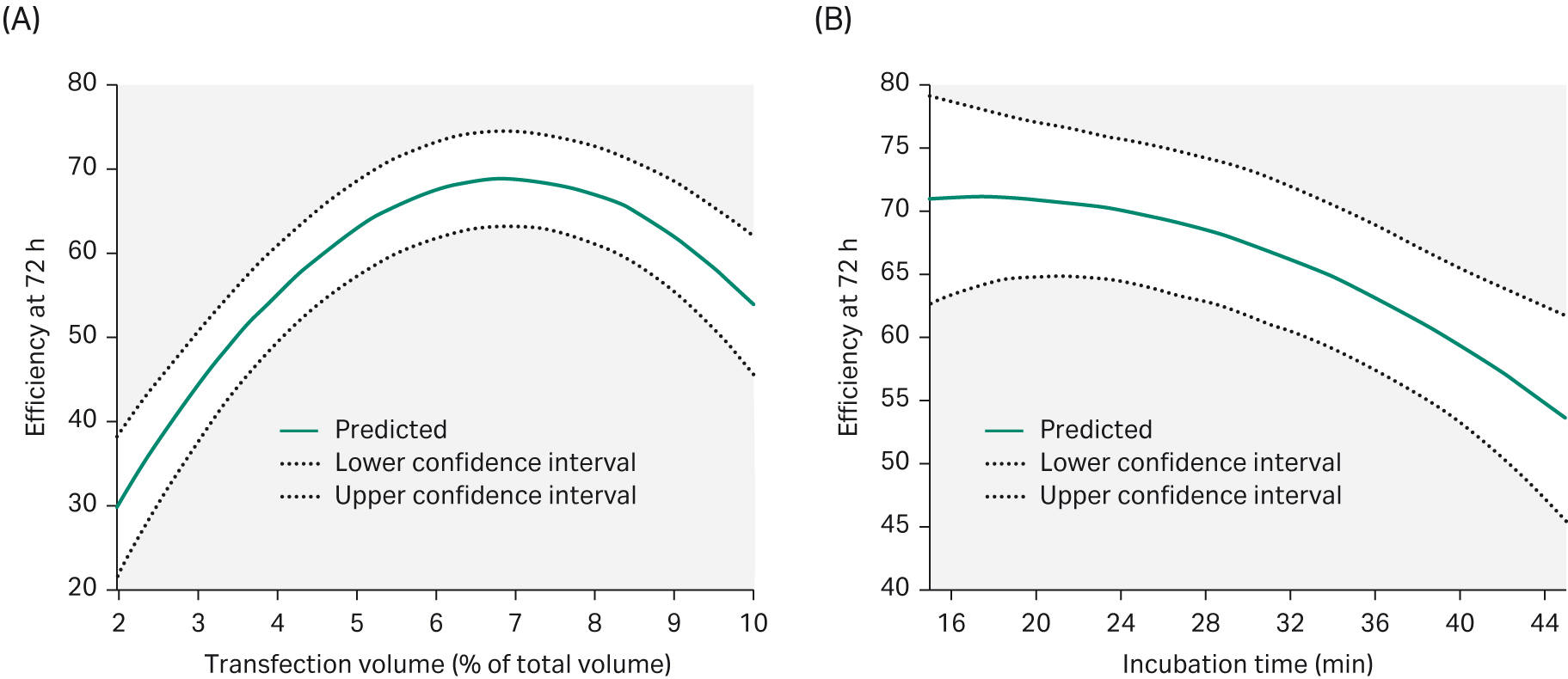
Fig 5. To achieve an optimal transfection efficiency after 72 h post-transfection, (A) the transfection volume needed to be 6% of the total volume. (B) Increasing the transfection incubation time would negatively affect transfection efficiency.
DoE2
For the second part (DoE 2), the following parameters were set based on the DoE1 evaluation. Temperature and DNA plasmid ratio were investigated and evaluated in DoE2.
The optimal amount of DNA varies depending on the characteristics of the transfected plasmid (e.g., size of plasmid, origin of replication). The dose of the various genes can also affect productivity and different ratios were tested for pAAV-RC2 Vector (a), pHelper (b), and pAAV-Vector-GFP (c). We also evaluated the effect on transfection efficiency at different temperatures during rAAV production — 34°C, 35.5°C, and 37°C.
The difference between DoE1 and DoE2 is marked with a circle in Figure 6.
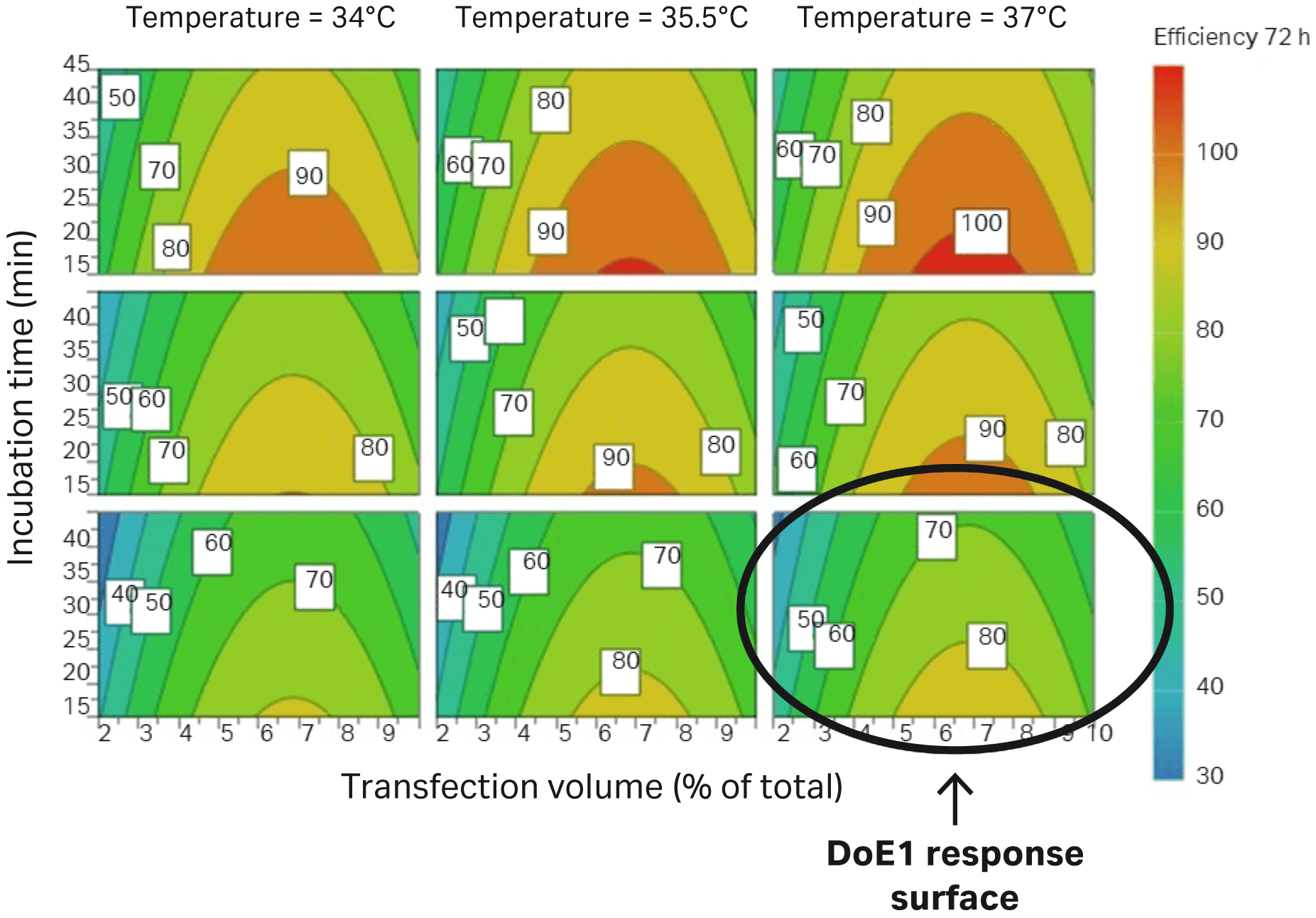
Fig 6. Parameters evaluated in DoE2 correlate to improved transfection efficiency in HEK293T cells (percentage of GFP-expressing cells) compared to DoE1, which is marked with a circle.
AAV infectivity and subsequent expression of GFP were determined by the transduction assay. Lysed samples were quantitated by transduction analysis and evaluated by flow cytometry, where transduced cells express the GFP-positive cells, and the results can be used to calculate infectious titers (TU/mL). This shows how the difference between DNA ratios improves the infectious titer.
The various parameters in DoE2 based on the transduction assay are shown in Figure 7. The circle shows the improvement between DoE1 and DoE2.
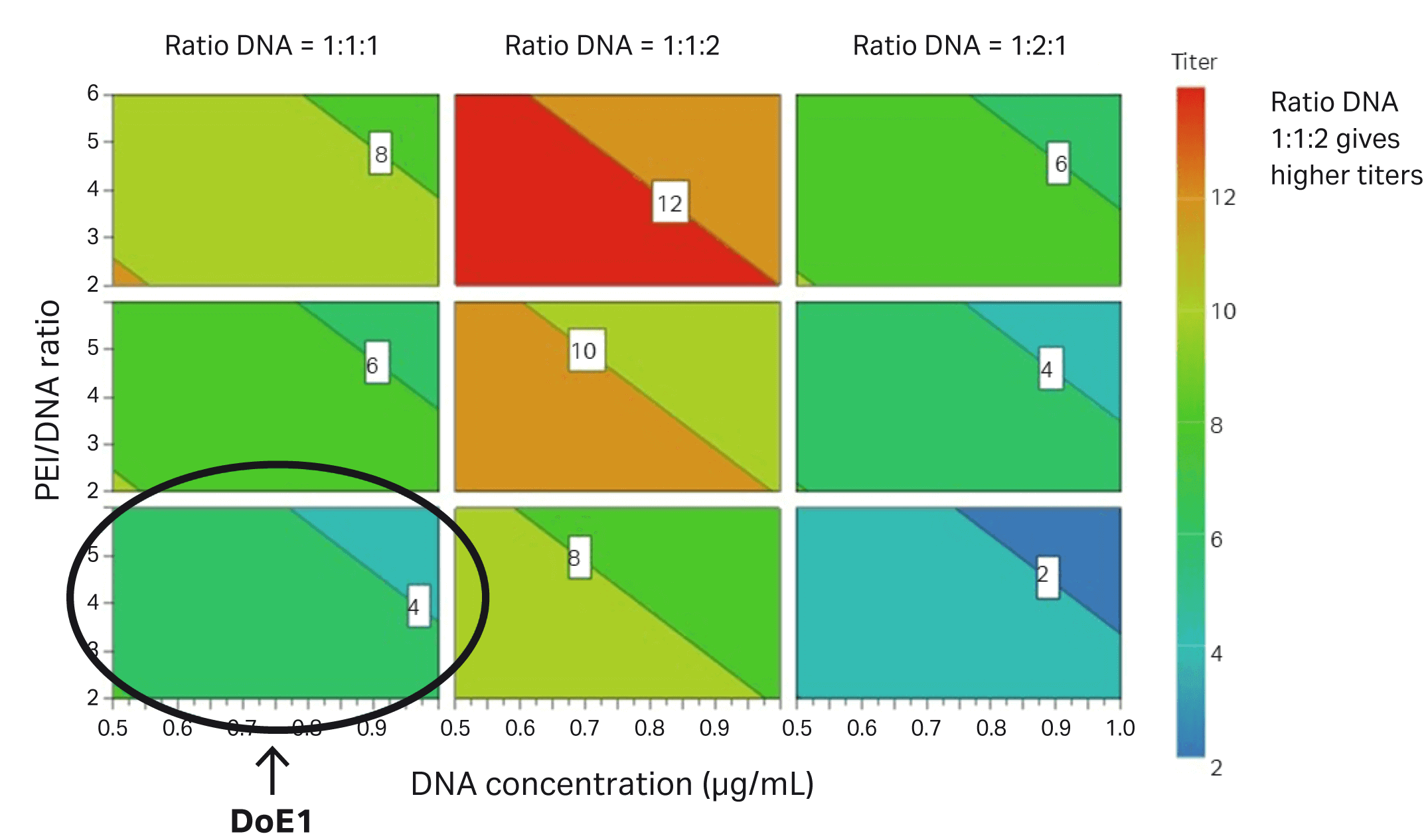
Fig 7. Parameters evaluated in DoE2 that contribute to improved infectious titer in HEK293T cells. The conditions from DoE1 are circled.
DoE3
qPCR allowed us to analyze the viral genome titer. This was important to further optimize the process and increase the share of full capsids in harvest material. The percentage of full capsids is estimated by comparing VG titer data by qPCR (VG/L) to the amount of capsid protein detected by ELISA (capsids/mL). It was evident that transfection efficiency showed a poor correlation with viral titer, measured by ELISA and qPCR.
Viral genome titer is improved when the VCD is increased to 1 × 106/mL.
DoE4
The percentage of full capsids and VG are the most important factors for an efficient separation of full capsids in the downstream purification process. We needed at least 10% of full capsids in the harvest to enable the rAAV2 purification process. Another DoE evaluation was therefore carried out on rAAV2, this time looking at the effect on DNA concentration and time of harvest (ToH) on viral genome titer and percent of full capsids during production.
We found that increased DNA concentration gives a higher VG titer but negatively affects the percentage of full capsids (Fig 8). To obtain a high VG titer but at the same time have 10% or more of full capsids, we found that a DNA concentration of 0.75 μg/mL would be needed.
Time of harvest has no significant effect on VG titer and percentage of full capsids under these conditions.
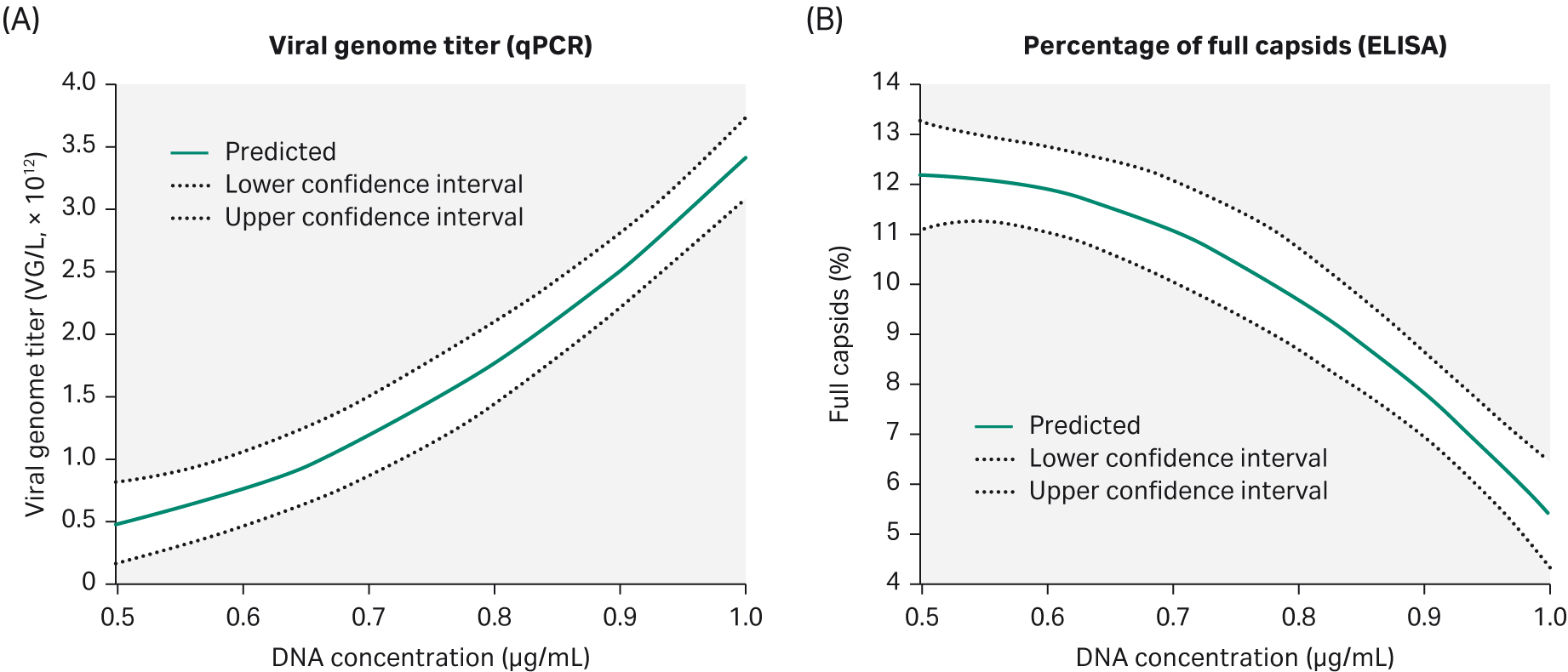
Fig 8. Parameters evaluated in DoE4 show that increasing the DNA concentration will give a higher (A) VG titer but the percentage of full capsids (B) is negatively affected.
Summarizing our experiments in DoE 1–4 for rAAV2, we see progress over time based on the studies for VG titer and percentage full capsids (Fig 9). However, our criterium for the percentage of full capsids was not fulfilled and it is generally accepted that AAV2 can be challenging to optimize for full capsids. Thus, we decided to test the identified conditions for AAV5, which is another clinically important serotype.
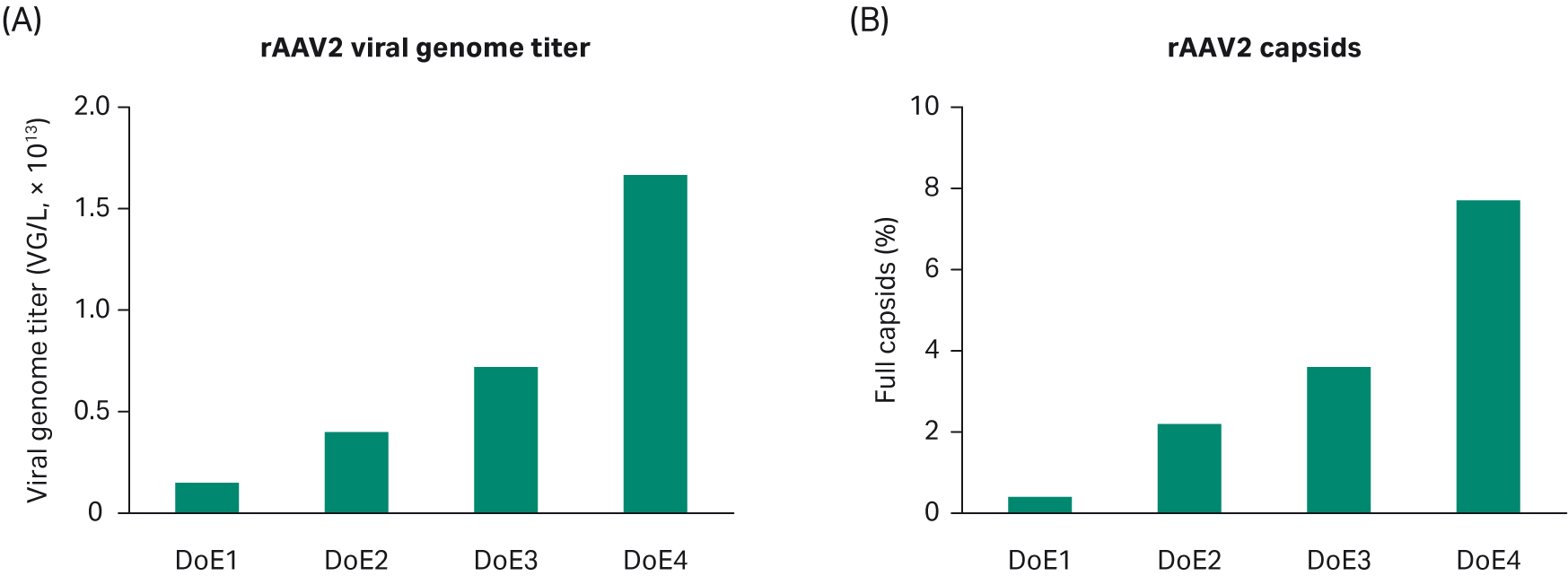
Fig 9. Progress over time based on the DoE studies for (A) VG titer and (B) percentage of full capsids.
DoE5
We verified that our AAV2 transfection protocol can be used for AAV5 production. Two parameters were important: DNA concentration and transfection incubation time. The results confirm the findings in DoE4: increased DNA concentration gives higher VG titer and a lower percentage of full capsids; transfection time has a minor effect on VG response (Fig 10).
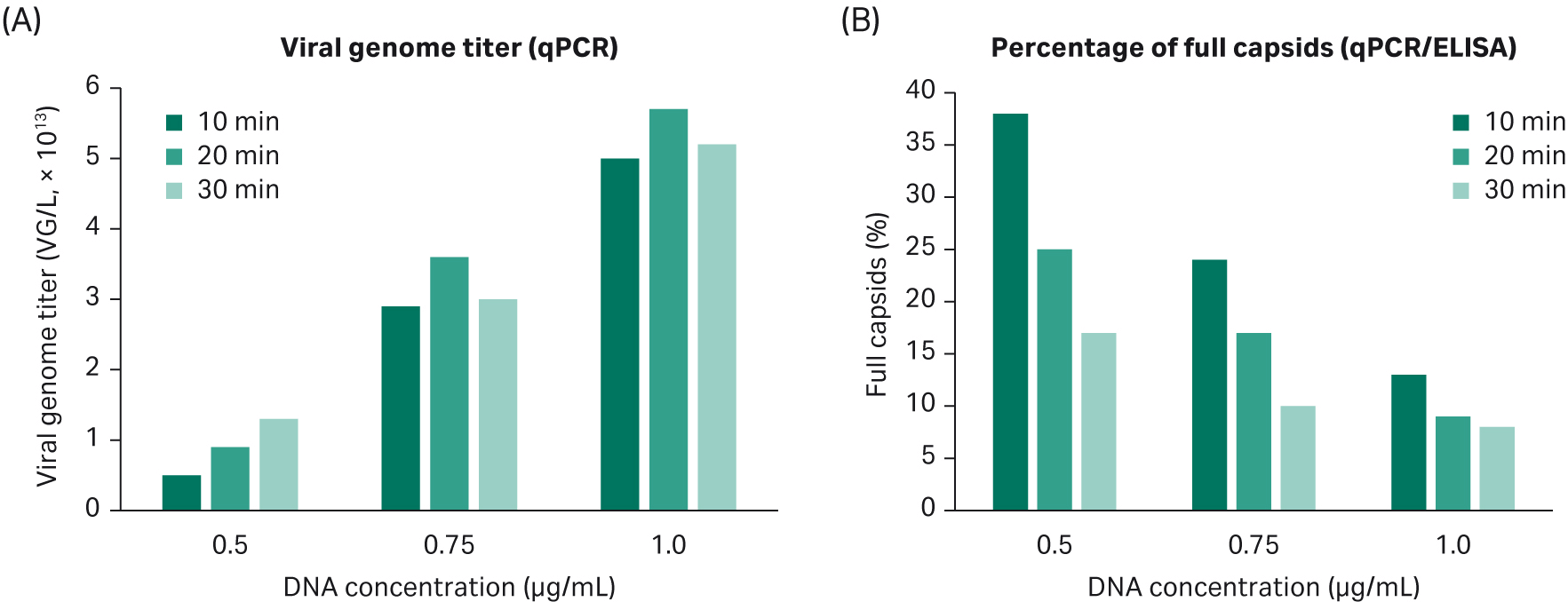
Fig 10. (A). Increasing DNA concentration gives a higher VG titer. Transfection incubation time has no significant effect on VG response. (B). Increased DNA concentration gives a lower percentage of full capsids. Transfection incubation time affects the percentage of full capsids more when using a low concentration of DNA.
We performed a design-space optimization model in our DoE where the probability of obtaining 10% full capsids or 15% full capsids was evaluated. The higher the percentage of full capsids, the smaller the area for the selection of transfection parameters (Fig 11).
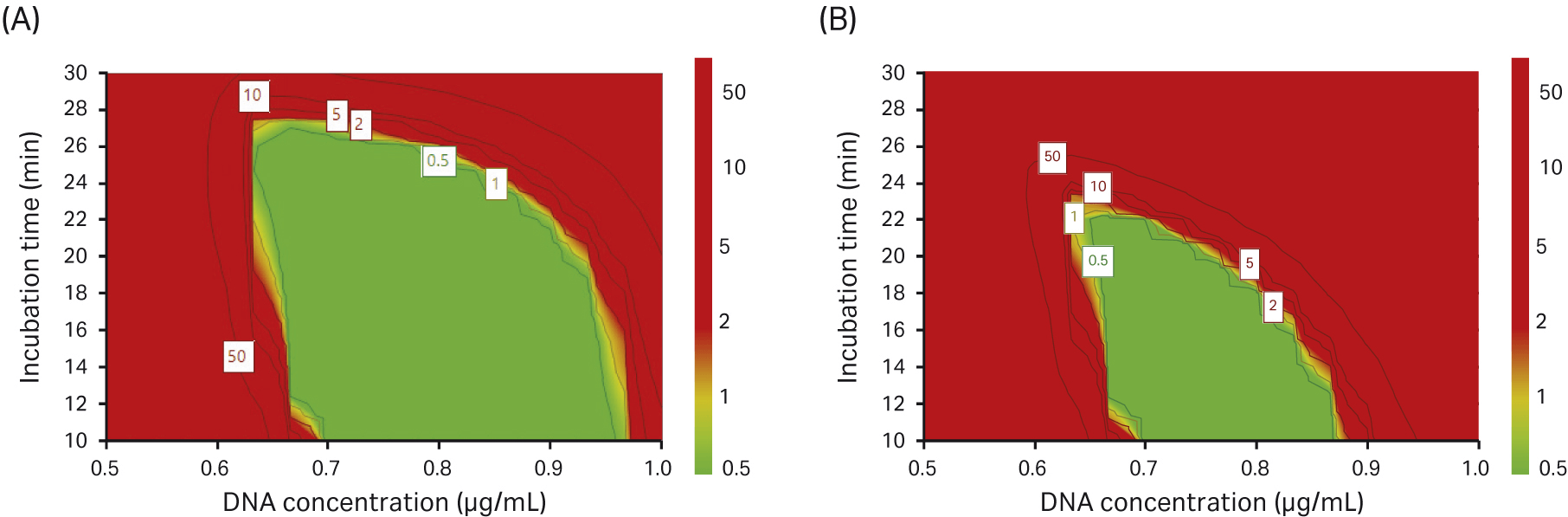
Fig 11. Design-space optimization where we investigated the probability of achieving (A) 10% full capsids and (B) 15% full capsids (green area).
In our DoE experiments, we found that a high concentration of DNA improved VG titer and transfection incubation time did not significantly affect the VG titer. However, when we looked at the percentage of full capsids, we saw the opposite effect; by reducing the DNA concentration and having a short incubation time, the percentage of full capsids increased.
In summary, the verification for rAAV5 production in the DoE5 study showed that a reduced transfection incubation time positively affected the percentage of full capsids. From the responses in the DoE experiments, we conclude that during transfection, a DNA concentration of 0.75 µg/mL and an incubation time of 15 min is likely optimal under the conditions tested.
Optimized protocol for transfection
The final transfection protocol and parameters are described in Table 2 below:
Table 2. Final parameters used for the transfection protocol
| Parameters |
rAAV |
|---|---|
| VCD (× 106/mL) |
1 |
| DNA concentration, total DNA (µg/mL) |
0.75 |
| DNA ratio: a:b:c |
1:1:2 |
| a | pAAV-RC5 Vector: 1 |
| b | pHelper: 1 |
| c | pAAV-GFP vector: 2 |
| PEI/DNA ratio (µL: µg) |
2:1 |
| Transfection volume |
5% of the total volume |
| Incubation time (min) |
15 |
| Temperature transfection |
RT |
| Temperature AAV production | 37°C |
| Time of harvest, ToH (h) | 72 |
To confirm the transfection protocol for rAAV5 production with 15 min transfection incubation time, we performed a 10 L production in ReadyToProcess WAVE™ 25 bioreactor. The harvest from this bioreactor run fulfilled all acceptance criteria for further purification in the downstream process.
Conclusions and discussion
We adapted HEK293T cells to serum-free suspension growth in HyClone™ media. Cells were efficiently adapted in a short time by direct transfer into the new medium. Cell viability was very high and with most cells in single-cell suspension. The cells grow rapidly in the HyCell™ TransFx-H medium with a population doubling time of approximately 20 h. Rapid cell growth will shorten the time needed for seed train scale-up and production.
During the adaptation, we saw that it was important to avoid very high cell densities as this could induce aggregation. In general, we kept the cells below 2.5 × 106cells/mL. Cell aggregates may reduce the transfection efficiency and cause problems in scale-up of the production. Importantly, we have also confirmed that HEK293 cells can also be adapted and cultured in HyCell™ TransFx-H medium with similar performance. The regulatory acceptance of HEK293T cells for production of AAV has been discussed due to the presence of the SV40 large T antigen and thus HEK293 cells are preferred in many cases.
We scaled up the production of rAAV5 material in ReadyToProcess WAVE™ 25, generating both high viral titers and high percentage full capsids in a scalable, robust process. Verification of the rAAV2 transfection protocol was performed to produce rAAV5 to achieve the optimal criteria for the plasmid purification:
- qPCR ≥ 1 × 1013 VG/L
- ELISA ≥ 1 × 1014 VP/L
- ≥ 10% full capsids
All the criteria for rAAV5 harvest material were met with these conditions.
Thus, we believe the optimized transfection protocol described in this work is suitable for several AAV serotypes and can be scaled up to volumes required for production of clinical grade AAV vectors. In this work we describe optimization for AAV 2 and 5, but this strategy has also been used successfully for AAV 8 and 9 (work in progress).
Materials and methods
Adaptation of HEK293T cells to a serum-free process for AAV production
Cryopreserved HEK293T adherent cells were thawed in T75 flask in HyClone™ Dulbecco’s Modified Eagle Medium (DMEM) with high glucose supplemented with 10% FBS (HyClone™).
We sub-cultivated up to a T225 flask and then transferred directly to respective new medium without serum, using 250 mL shaker flask with 40 mL medium and a cell density between 0.2 to 0.3 × 106 cells/mL depending on the number of days between splits. The cells were cultured in a shaking incubator at 135 rpm (radius 50 mm) at 37°C and 5% CO2.
Cell culture adaptation protocol
Direct adaptation was performed according to the following step-by-step protocol. The cell culture medium was supplemented with 0.1% Pluronic™ F-68 (0.1%) nonionic surfactant (Thermo Fisher™ Scientific) to reduce aggregates and shear stress.
- Adherent cells were detached and centrifuged at 150 × g for 5 min. Cells were resuspended by thorough mixing in a 5 or 10 mL pipette approx. 20 times.
- Cells were seeded at 0.3 × 106 cells/mL to 40 mL in a 250 mL shake flask. The volume during adaptation was kept low to ensure good culture mixing.
- Cells were split every second day. The pellet was thoroughly resuspended to keep the viable cell density (VCD) below 2.0 to 2.5 × 106/mL to avoid aggregates. A 40 µm mesh was used during filtration to remove aggregates.
- VCD and morphology were monitored and NucleoCounter™ NC200™ Automated Cell Counter (Chemometec) was used for counting aggregates.
- A Working Cell Bank (WCB) was created after 10 passages using 15 × 106 cells/vial.
Note that an important step in the adaptation is how you seed your cells and ensure good cell culture mixing. To avoid aggregates, we used Pluronic™-F68 from the beginning of the adaptation and kept the cell density below 2.5 × 106 cells/mL. For optimal cell culture results, take the effect of culture volume in relation to shaking flask size into account.
Cell banking
HEK293T cells were cryopreserved in a 1:1 ratio of fresh cell culture medium and freeze medium (HyClone™ brand HyCryo 2x cryopreservation medium) by pelleting cells after centrifugation for 5 min at 150 × g, RT.
The cell pellet was resuspended in a fresh cell culture medium before the addition of an equal volume of freeze media. Aliquots of 1 mL/cryotube containing 15 × 106 cells/mL were frozen in CoolCell™ (TATAA Biocenter) at -70°C overnight and transferred to a -150°C freezer for long-term storage.
Cell culture
For the cell-growth comparison and maintenance culture, cells were thawed and seeded and in a 125 mL volume with a 25 mL cell culture medium. The cells were sub-cultivated two times a week with a seeding density of 0.2 × 106 cells/mL. Cell culture was performed in 250 mL shake flasks with a working volume of 40 mL. Cells were counted using Vi-CELL™ cell analyzer (Beckman Coulter) and seeded in a new shake flask with fresh medium (E125/E250 flasks). The medium was supplemented with 4 mM L-glutamine and 0.1% Pluronic™ F-68.
Plasmid preparation and purification
The three plasmid constructs for transfection in this study — pAAV-GFP control vector, pHelper vector, and pAAV-RC vector— were sourced from Cell Biolabs. The constructs were prepared for transfection, sequenced, amplified in bacteria, and purified using a plasmid purification kit.
The plasmid was purified using either EndoFree™ Mega Plasmid Kits (Qiagen™) or Macherey-Nagel™ NucleoBond™ PC 10000 EF kits. For specific procedures, see supplier instructions. Each plasmid pellet was dissolved in 0.75 mL sterile dH2O.
We evaluated plasmid quality using restriction enzyme digestion and the fragments were analyzed on an agarose gel.
An overview of preparation using Luria broth (LB), shake flasks, and purification kit is shown in Figure 12.
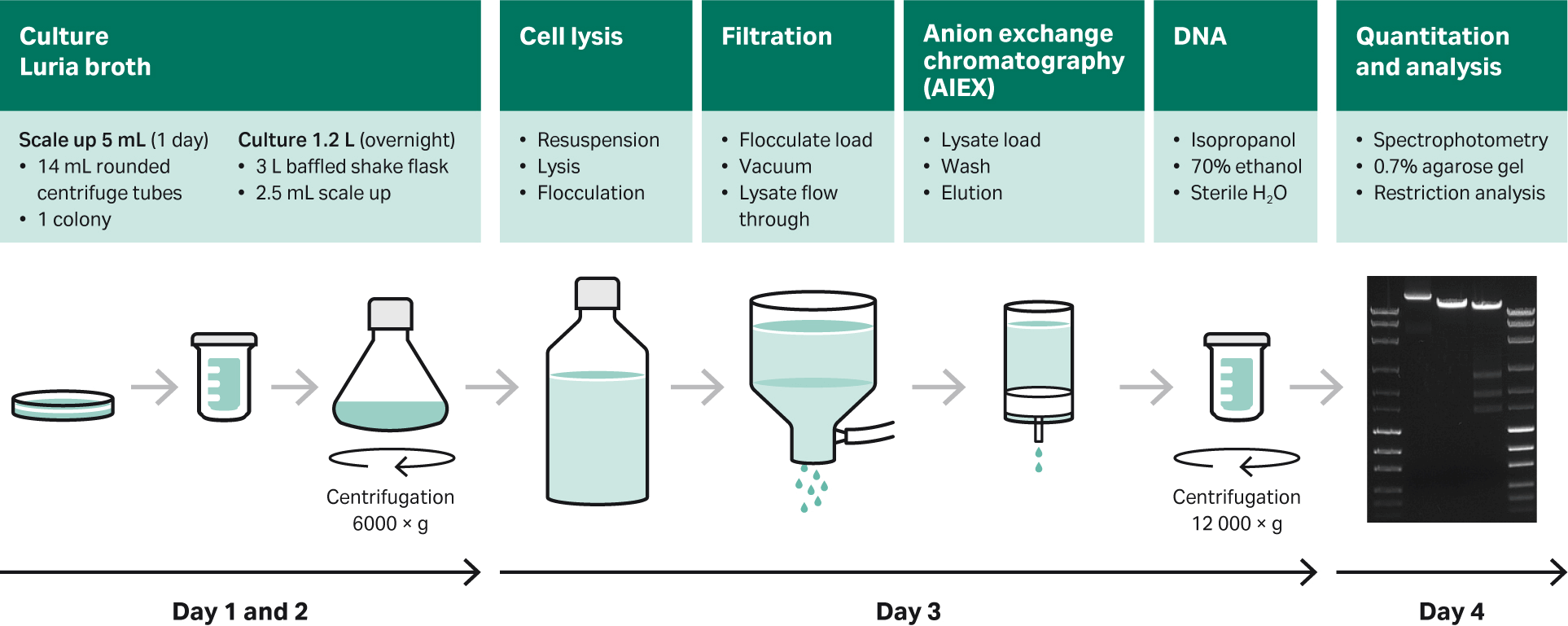
Fig 12. Overview of preparation using LB, shake flasks, and purification with commercially available purification kits.
Transfection of HEK293T suspension cells in shake flasks
Day 0:
- Seed cells at 1 × 106 cells/mL. Make sure that maintenance cells are in good condition based on cell growth and aggregates. Ensure good control of the cell passage number and do not use cells over 40 cell passages.
- Calculate and normalize plasmid amounts for transfection. For shake flask (20 mL) culture to be transfected, prepare 0.75 μg/mL of total DNA in serum-free medium to a final volume of 5% of the final volume in a 50 mL centrifuge tube and mix by pipetting. For larger cultures, use larger transfection tubes/containers. Note: Only use plastics made of polypropylene, not polystyrene!
- Prepare PEI Max (Polysciences) solution (1 mg/mL), mix before use.
- Add x μL of PEI (for a 1:2 DNA:PEI ratio) to the DNA mixture that was prepared in Step 1, (for a 20 mL culture, this would be 15 µg DNA and 30 µL PEI). Mix the solution by flicking the tube or swirling the bottle back and forth gently.
- Incubate for 15 min at room temperature. Add the PEI/DNA mixture slowly to the cells and mix by gently swirling the flask.
- Return the flasks to the cell culture incubator (37°C, 5% CO2).
Day 3: 72 h post-transfection
- Collect samples and check transfection efficiency with flow cytometry.
- Lyse the cells in the shake flask by adding 10% of lyse buffer (1.65 M NaCl, 5.5% Tween™ 20, 11 mM MgCl2) to the culture and incubate at 37°C with mixing for 20 min (return the shake flask in the CO2 incubator).
- Add 40 U DNAase/mL (Denarase™, c-LEcta GmbH) to the lysed culture and incubate at 37°C for at least 4 h or overnight in a CO2 incubator.
- Centrifuge lysed culture at 850 × g for 10 min, aliquot the supernatant, and store at -80°C. Discard the pellet.
Evaluation of rAAV2 production process using design of experiments (DoE)
We used DoE methodology to optimize the plasmid transfection protocol for rAVV production in the HEK293T suspension cell system for the HyCell™ TransFx-H medium. We could evaluate the impact of 50 different conditions/parameters on rAAV transfection. DoE was analyzed and evaluated using MODDE™ Pro v12.0.1 software (Sartorius Stedim GmbH).
The DoE was set up in different parts. We started to investigate the effects of plasmid DNA concentration, different cell densities, PEI/DNA ratio, transfection volume, and incubation time. We continued with fixed parameters from the previous DoE and added new parameters to the next DoE. Evaluation of temperature, as well as DNA plasmid ratio, were investigated and evaluated.
Transfection efficiency and transduction assays detecting GFP-expressing cells were evaluated by flow cytometry (BD Biosciences). Viral genomes (VG, full capsids) were determined by qPCR (VG/L) and the total viral particle titer (VP/L) was determined by ELISA (Progen). The ratio of qPCR:ELISA was used to estimate the percentage of full capsids.
A summary of five DoE studies, where we achieved the fully optimized conditions in DoE5, is shown in Table 3.
| Parameters |
DoE1 (AAV2) |
DoE2 (AAV2) |
DoE3 (AAV2) |
DoE4 (AAV2) |
DoE5 (AAV5) |
|---|---|---|---|---|---|
| VCD (× 106/mL) |
0.75 |
0.75 |
1 | 1 | 1 |
| DNA concentration (µg/mL) |
0.75 |
0.75 |
0.75 |
0.75 |
0.75 |
| DNA ratio (pAAV-RC:pHelper:pAAV-GFP) |
N/A |
1:1:2 |
1:1:2 |
1:1:2 | 1:1:2 |
| PEI/DNA ratio (µL:µg) |
2:1 |
2:1 |
2:1 |
2:1 |
2:1 |
| Transfection volume (% of total volume) |
6 | 6 |
5 | 5 | 5 |
| Incubation time (min) |
30 | 30 |
20 | 20 |
15 |
| Temperature |
RT |
RT |
RT |
RT |
RT |
| Temperature AAV production (°C) |
37 | 37 |
37 |
37 |
37 |
| Time of harvest, ToH (h) |
72 | 72 |
72 |
72 |
72 |
| Analytical result |
|||||
| Infectious titer (TU/mL) |
7.0 × 105 |
1.4 × 106 |
N/A |
N/A |
N/A |
| ELISA (VP/L) |
8.0 × 1012 |
4.4 × 1012 |
5.2 × 1013 |
1.2 × 1013 |
2.1 × 1014 |
| qPCR (VG/L) |
N/A |
N/A |
3.6 × 1012 |
1.3 × 1012 |
3.7 × 1013 |
| Full capsids (%) |
N/A |
N/A |
7 | 10 | 18 |