Scalable, start-to-finish process for rAAV type 5 production
We developed a production process for a recombinant adeno-associated virus (rAAV), from upstream cell culture to downstream purification, using modern tools and technologies. To demonstrate scalability of the upstream process, virus production in suspension-adapted cells was performed by triple plasmid transfection in a stirred-tank bioreactor. The downstream process was carefully optimized to maximize the proportion of full capsids and to meet stringent regulatory demands on product purity and quality. Novel analytical assays were used in parallel with established techniques for comparison and to ensure accurate monitoring of the processed material. The process is easily scaled and GMP compatible with both single-use and hard-piped process equipment.
Introduction
Human embryonic kidney 293 (HEK293) cells are susceptible to a broad range of viruses and can be easily transfected and are thus frequently used in the production of viral vectors and vaccines. Here, suspension adapted HEK293T cells and optimized triple plasmid transfection were used to produce rAAV, the main vector for gene therapy.
The key for a successful upstream production is having consistently high full capsid productivity, preferably using animal component-free suspension cell culture media. Important for the downstream process are efficient steps with good recoveries producing a high proportion of full capsids and low levels of impurities in the final bulk product. Scalable, cost-efficient, and robust upstream production, filtration, and chromatography-based processes are required for the purification of rAAV.
Depending on the clinical indication and administration route, the dose requirements and production volumes can vary significantly. Local administration requires a smaller dose than systemic administration. Hence, the interest in scalable platforms for industrial rAAV production is increasing. Here, we present results from a start-to-finish process for rAAV5 serotype, including virus production, harvest by cell lysis, clarification, concentration and buffer exchange, affinity capture, and finally anion exchange polishing to enrich or purify the full capsids (Fig 1).
We have used several analytic methods for titer, the proportion of full capsids, and impurity assessment. A new Biacore™ surface plasmon resonance (SPR) assay for robust and accurate total rAAV5 titer determination was developed and used. A more detailed description of the process development work and Biacore™ assay development are explained in detail separately.
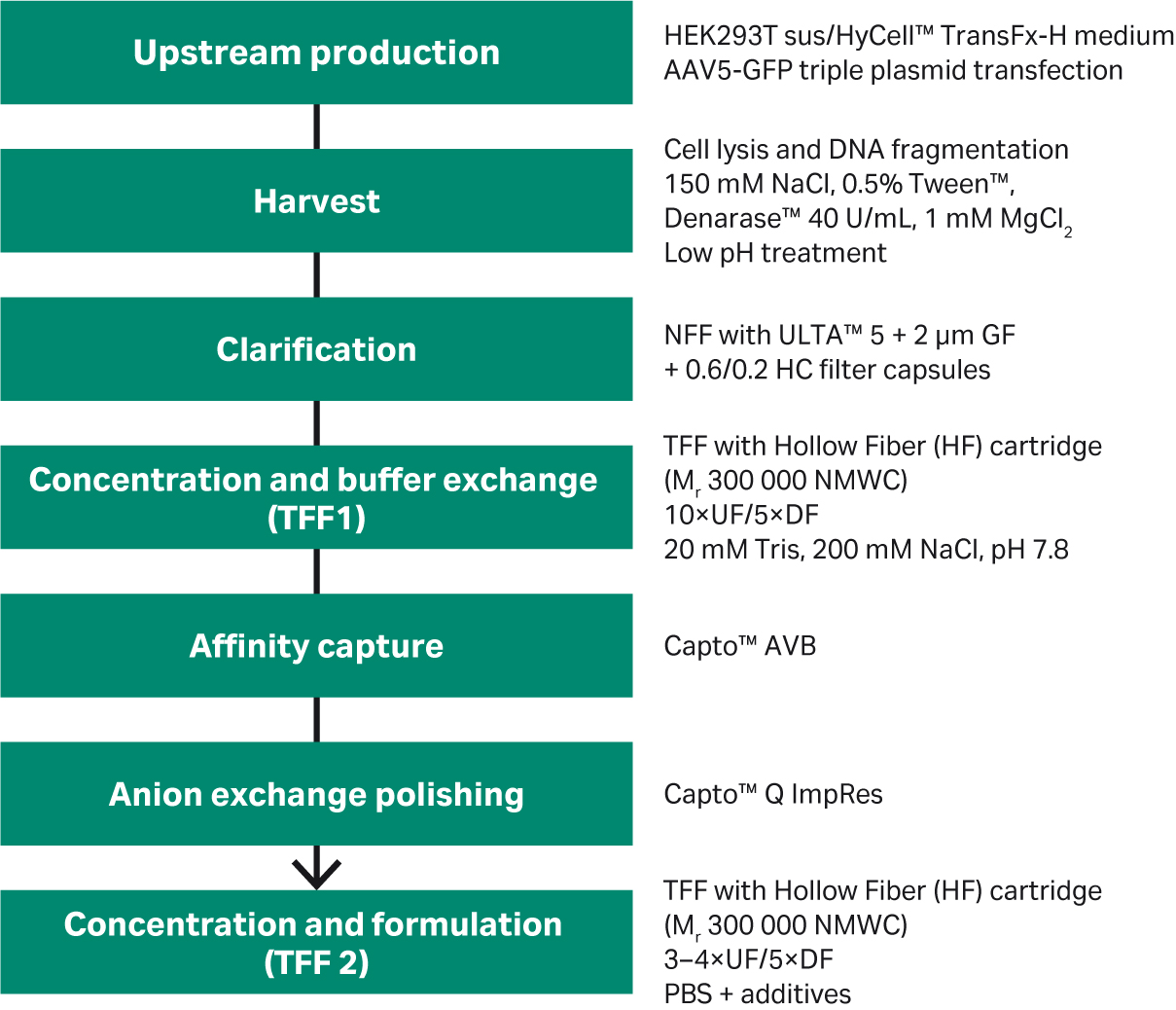
Fig 1. rAAV5 production process overview.
Materials and methods
rAAV production
HEK293T adapted to suspension culture (CRL-3216 ATCC) were grown in HyClone™ transfection medium, HyCell™ TransFx-H medium (Cytiva), using Xcellerex™ XDR 10 bioreactor. We used an optimized triple plasmid transfection protocol to produce rAAV5-GFP in triplicate at the 10 L scale. The viable cell density at transfection was 106 cells/mL, total DNA concentration was 0.75 µg/mL with plasmid ratio of 1:1:2 (Rep/cap: helper: transgene GFP), PEI/DNA ratio was 2:1 (µL:µg). The transfection volume was 5% of the total volume and the time of transfection was 15 min at RT. We harvested cells at 72 h after transfection.
Harvest and clarification
At the time of harvest, we lysed the cells by adding 1/10 volume (1 L) of lysis buffer containing 5.5% Tween™ 20, 1.65 M NaCl, and 11 mM MgCl2 directly into the bioreactor. After 20 min of lysis, the harvest was treated with 40 U/mL of Denarase™ (c-LEcta) to degrade the DNA by incubation in the bioreactor for 4 h at 37°C.
To minimize issues with spontaneous precipitations, the pH was lowered to between 4.0 and 4.2 by adding roughly 1/100 volume of 1 M citric acid stock solution, followed by stirring for 20 min to induce precipitation. Finally, the pH was increased to between 7.0 and 7.5 by the addition of roughly 1/30 volume of 1 M Tris base. The resulting precipitate was allowed to sediment and was discarded. The cell lysate was clarified by normal flow filtration (NFF) using a combination of 5 µm GF (10 in., 0.53 m2), 2 µm GF (10 in., 0.53 m2), and 0.6/0.2 µm HC (10 in., 0.55 m2) ULTA™ filters. The filters were prewetted with 20 mM Tris pH 7.8, 200 mM NaCl before use. The lysate was applied at approximately 200 mL/min and the pressure did not exceed 0.01 MPa (1 bar, 14.5 psi).
Concentration and buffer exchange (TFF1)
Concentration and buffer exchange was performed by tangential flow filtration (TFF) on two ReadyToProcess™ hollow fiber filters (UFP-300-C-4x2MA) connected in parallel with a filter area of 2800 cm2 and an NMWC of Mr 300 000 at a shear rate of 9000 s-1 and transmembrane pressure (TMP) of 0.09 MPa (0.9 bar, 13.1 psi) using ÄKTA flux™ 6 filtration system. A 10-fold ultrafiltration/5-fold diafiltration step was conducted to reduce volume, impurities, and exchange the buffer to 20 mM Tris pH 7.8, 200 mM NaCl to prepare the rAAV-containing sample for subsequent downstream purification steps.
Affinity capture
The rAAV affinity capture was performed using a 5 mL Capto™ AVB HiTrap™ chromatography column. Columns were operated on an ÄKTA pure™ 25 chromatography system and the flow velocity of 150 cm/h equivalent to 1 min residence time for all steps using the method outlined in Table 1. After the low pH elution, the sample was neutralized by the addition of 1/10 volume of 1 M Tris-HCl, pH 9.0.
Table 1. Method for Capto™ AVB affinity capture step

Polishing for full capsid enrichment
The neutralized Capto™ AVB eluate was diluted 10-fold in an equilibration buffer without MgCl2 (A buffer) to a conductivity of between 1 to 3 mS/cm. A Capto™ Q ImpRes HiScale™ column with a column volume of 51 mL and a bed height of 10 cm was used. Columns were operated on an ÄKTA pure™ 25 system and the flow velocity of 250 cm/h or 2.4 min residence time for all steps, using the method outlined in Table 2. Following load and wash the bulk of the empty capsids were eluted with buffer A, after which the full capsids were eluted over a linear gradient to 20% B. The polishing step of run 1 was a re-run, since the first run had too high an MgCl2 concentration at the elution hold step with buffer A, resulting in both full and empty capsids in the flowthrough. The material was repurified on Capto™ AVB before being reloaded on Capto™ Q ImpRes resin with the optimal MgCl2 concentration.
Table 2. Method for Capto™ Q ImpRes polishing step

Concentration and formulation (TFF2) and sterile filtration
The pooled fractions from the Capto™ Q ImpRes step were concentrated to approximately 100 mL by tangential flow filtration (TFF) using hollow fiber with a filter area of 140 cm2 (UFP-300-C-MA) and an NMWC of Mr 300 000 at a shear rate of 7000 s-1 (220 mL/min) and a TMP of 0.08 MPa (0.8 bar, 11.8 psi) using ÄKTA flux™ S system. The retentate was then formulated by five diafiltration volumes of PBS supplemented with 1% sucrose and 0.1% poloxamer-188 and the final retentate was concentrated down to a target concentration of 1 × 1012 VG/mL. Process aliquots of the final bulk were filtered with a small-scale syringe filter (PES, 0.2 µm).
Analytics
To measure virus titer, total virus particles were determined by ELISA (Progen) or SPR (Biacore™), viral genomes (VG) by qPCR targeting the CMV promoter sequence, and functional titer was determined by a transduction assay using a cell-based assay followed by flow cytometry. To measure impurities, total protein was analyzed using a Micro BCA assay and total DNA was analyzed by Quant-iT™ PicoGreen™ dsDNA Assay (Thermo Fisher Scientific). Host cell DNA (hcDNA) was analyzed by qPCR targeting HEK293 GAPDH sequence and HCP was analyzed using an ELISA (Creative Diagnostics). Cryo-TEM analysis was used to characterize rAAV5 and performed by Vironova AB.
Results and discussion
Upstream rAAV5 production
We have previously described how a generic transient transfection protocol for the production of different adeno-associated virus serotypes was developed. This was further optimized and developed into a scalable suspension cell culture process with animal-derived component-free medium producing high rAAV titers (for details on the cell culture and transfection optimization see here).
Adherent HEK293T/HEK293 cells were adapted to suspension culture in the HyClone™ transfection medium, HyCell™ Transfx-H. The transfection conditions and parameters were optimized using the Design of Experiments (DoE) methodology.
Suspension cells were expanded from small cell culture (20 mL) in shake flasks up to a 10 L culture in a benchtop Xcellerex™ XDR 10 single-use bioreactor system. rAAV was produced at high titers upon transient plasmid transfection directly in the bioreactor. The three 10 L runs of suspension HEK293T cells triple plasmid transfected for rAAV5 production using the previously optimized protocol (Table 3) showed similar cell growth and viability (Fig 2).
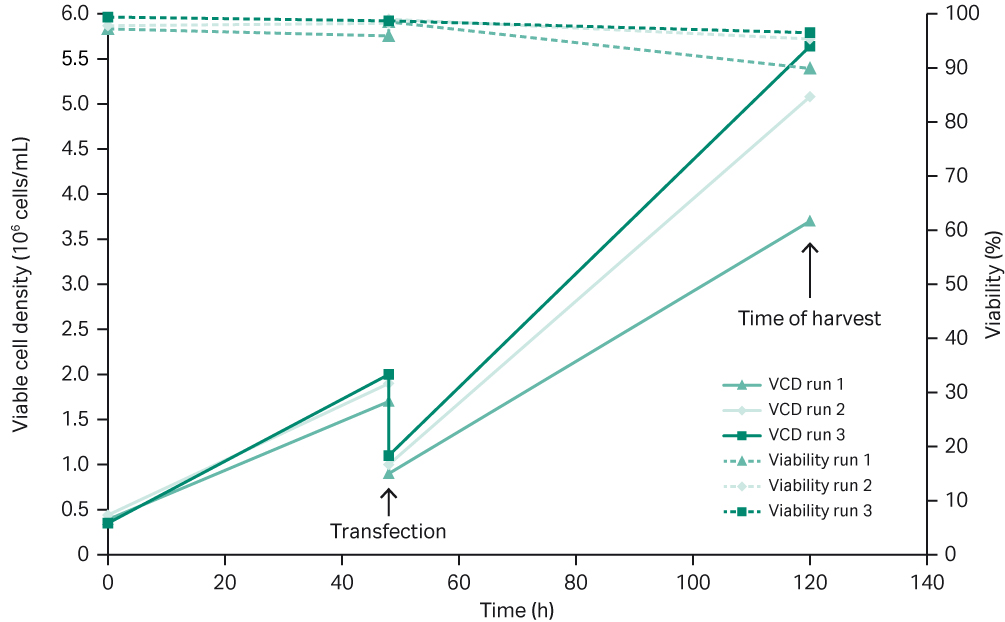
Fig 2. Three 10 L runs of rAAV5 production using Xcellerex™ XDR 10 benchtop bioreactor.
The target titer of total rAAV5 viral particles (VP) was ≥ 1 × 1014 VP/L and viral genomes was ≥ 1 × 1013 VP/L with more than 10% full capsids were reached for all three runs (Fig 3). Thus, the transfection-based rAAV production is robust at the 10 L scale and suitable for further scale-up. The material from these cultures was used for the purification of rAAV as described below.
Table 3. Optimized transfection protocol

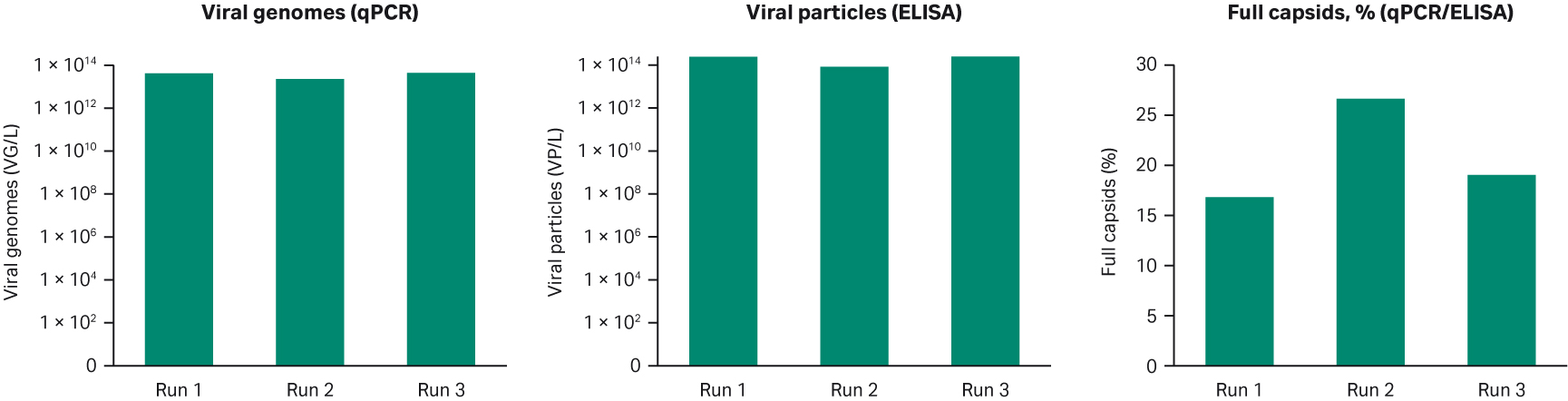
Fig 3. Productivity of three 10 L runs of rAAV5 production.
Downstream purification
The rAAV5 was released by lysing the host cells by the addition of Tween™ 20 detergent directly in the bioreactor. After nuclease treatment, the feed was clarified using three normal flow filters followed by a TFF step with hollow fiber with 300 000 MWCO for a 10-fold concentration and buffer exchange with five diafiltration volumes. We compared the performance using hollow fiber with 100 000 MW or 300 000 MWCO and we had similar virus recovery but improved total protein and total DNA removal using the 300 000 MWCO (data not shown).
The main advantage of the TFF step is that the reduced volume significantly decreases the loading time for the affinity capture step with Capto™ AVB. The total recovery of rAAV5 particles was around 70% for all three runs after the TFF1 step. The retentate was run through a Capto™ AVB affinity column which provided an approximately 100-fold concentration and an efficient purification with recovery of between 70% to 95% total rAAV5 particles in the eluate peak (Fig 4). The previously optimized protocol seen in Table 1 was used. The elution buffer used was glycine pH 2.7 without NaCl, which was shown to be critical for rAAV5. The absence of salt in the elution buffer did not cause any aggregation of rAAV5 from analytic size exclusion chromatography with Superose™ 6 Increase (< 1% data not shown). The binding capacity was determined to be between 2 to 5 ×1014 VP/mL resin.
(A)
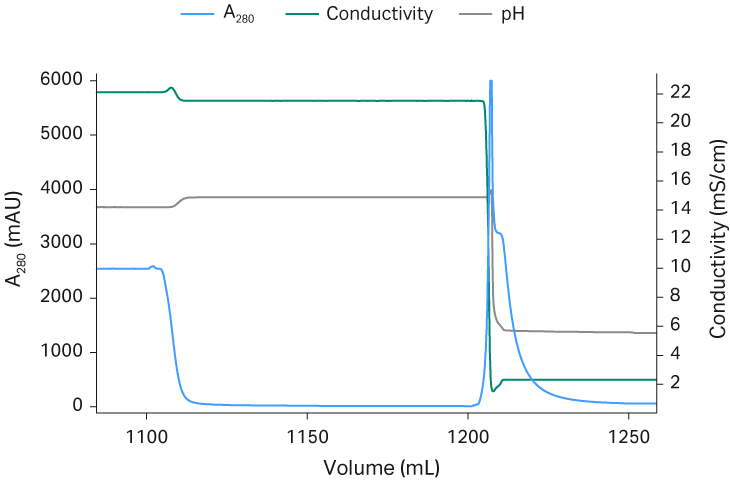
(B)
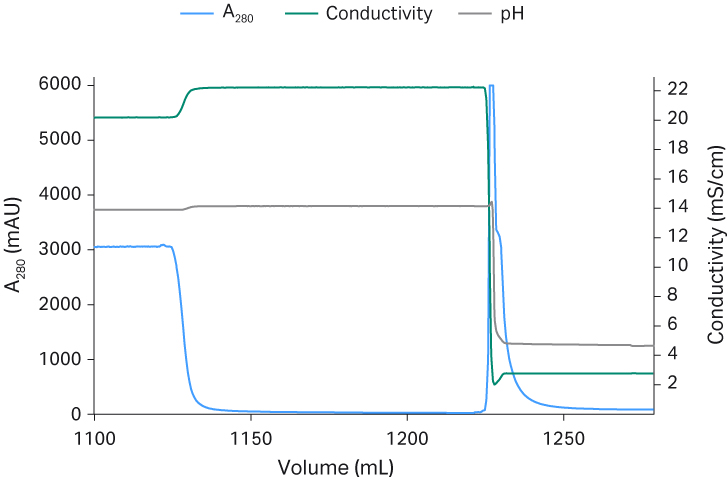
(C)
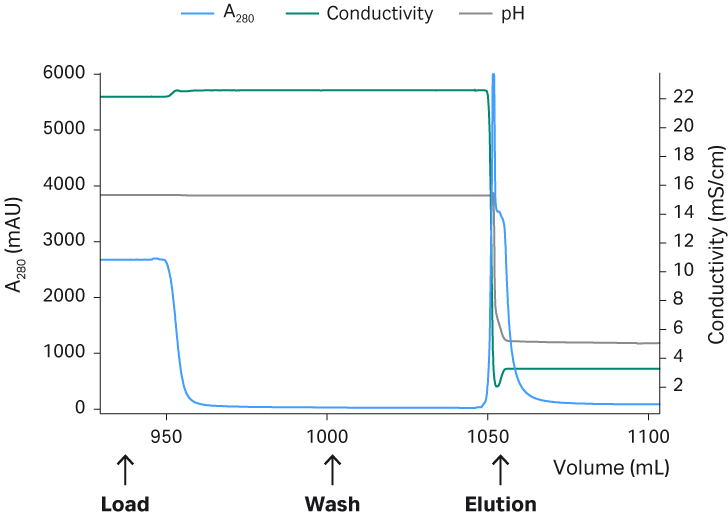
Fig 4. Capto™ AVB chromatograms from process run 1 (A), run 2 (B), and run 3 (C).
Since the affinity capture will not discriminate between full and empty capsids, the purpose of the polishing step is to reduce or remove the empty capsids as much as possible. Since there is a small difference in pI between full and empty capsids, with the full capsid having a lower pI, an anion exchange step can be used to separate the two capsid forms. The development of the polishing step with Capto™ Q ImpRes chromatography resin is described in detail here. Capto™ Q chromatography resin with dextran extenders is also a good alternative for enhanced full and empty capsid separation.
The Capto™ AVB eluate was neutralized and diluted down with equilibration buffer to low conductivity before application to the Capto™ Q ImpRes column to ensure rAAV5 binding. After a wash, most empty capsids could be eluted first using an isocratic step with equilibration buffer containing 18 mM MgCl2. A shallow NaCl gradient was applied (keeping the MgCl2 concentration constant) and most of the full capsids eluted. The fractions with the highest percent full capsids were selected and pooled. Since the separation of full and empty was not complete, a pooling strategy was employed, balancing the percent of full capsids versus the recovery of viral genomes, resulting in a recovery with 60% viral genome particles The polishing runs were reproducible as shown in the chromatograms in Figure 5. The Capto™ Q ImpRes pool was concentrated and formulated by a second TFF step and finally sterile filtered using a 0.2 PES filter.
The results from the analysis of the final samples show that we reached similar titers and regulatory purity targets for host cell DNA and protein per dose of 1 × 1011 VG in all three runs. We achieved approximately 3 to 4-fold enrichment of full capsids from an initial average of 15% by qPCR:ELISA in the harvest material to between 42% and 71% full capsids in the final sample, depending on analysis method (Table 4). While determination of the percentage of full capsids using qPCR:ELISA relies on two independent assays, both with a significant inherent variability, which resulted in higher uncertainty compared to other methods.
We also used analytical anion exchange and calculated the percent full capsids based on peak area (Table 4). Since it was a single analysis for both empty and full capsids there should be increased accuracy. We recommend using at least one additional orthogonal analytical method for percent full capsids to confirm data and increase accuracy. We also performed the cryo-TEM analysis of our samples before and after the polishing step, and we reached a similar fold increase (data not shown). The full and empty capsids analysis is a challenge and there are many different possible methods to try but the accuracy depends on the method used (1).
(A)
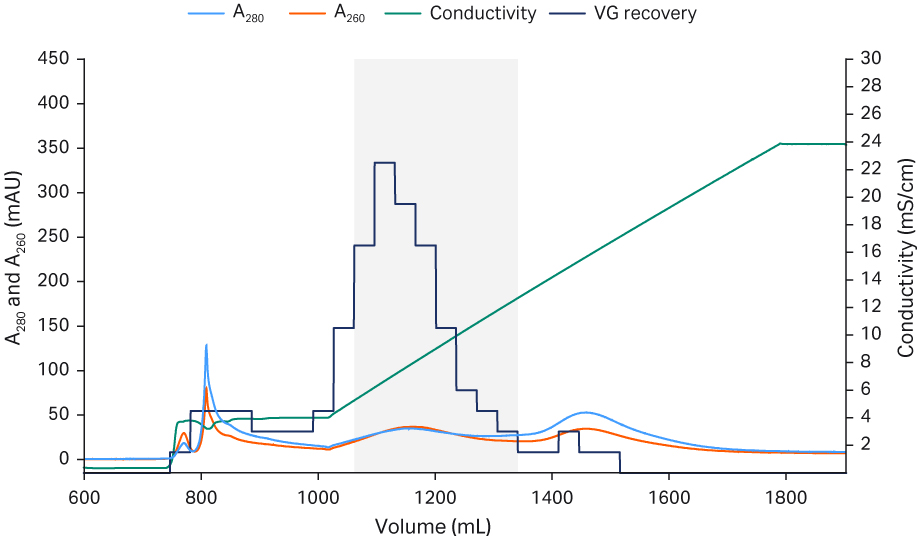
(B)
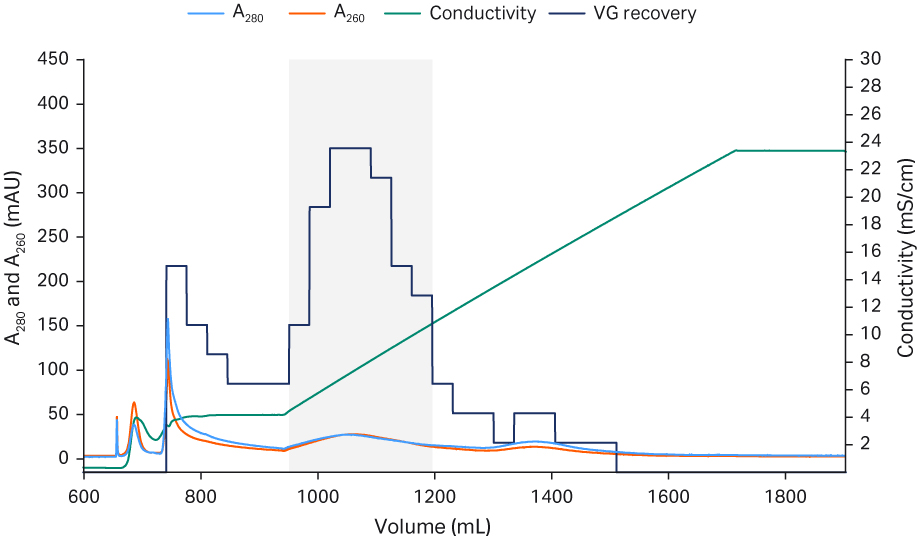
(C)
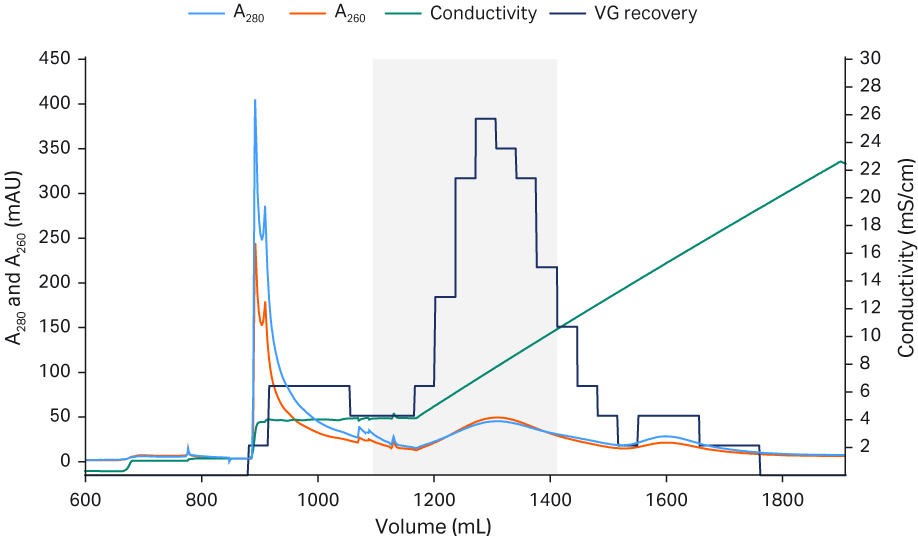
Fig 5. Chromatograms from Capto™ Q ImpRes polishing process run 1 (A), run 2 (B), and run 3 (C) The pooled fractions containing the highest percent full capsids and sufficient viral genome recovery are indicated by the gray box.
The Capto™ Q ImpRes pool containing the full capsids was concentrated 4- to 5-fold to target approximately 1 × 1012 VG/mL and formulated into 100 mL final volume PBS buffer containing additives in a second TFF step. The samples were sterile filtered and analyzed for total particle titer with both ELISA and Biacore™ titer assay and viral genomes by qPCR. Overall, the three processes showed good repeatability with similar titers and yields of the final samples (Fig 6 and Table 4). From the 10 L harvest material, approximately 1 × 1013 VG/L to 1 × 1012 VG/L harvest was obtained. This means that from each liter of cell culture supernatant between 600 to 1400 doses could be produced (depending on dose size).
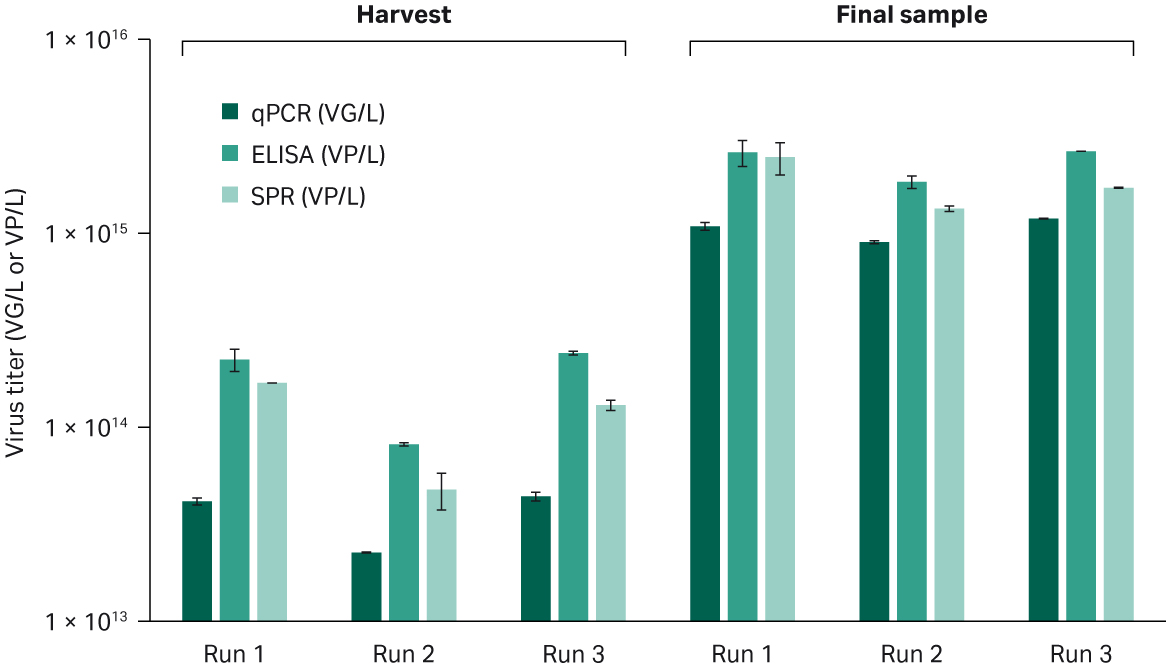
Fig 6. rAAV5 titers in harvest material and final purified samples from run 1, 2, and 3 determined by ELISA and SPR with Biacore™ (VP/L), or qPCR (VG/L).
The recoveries from each step were between 60% and 100% but the overall process yield was around 25% and the largest losses came from the polishing step (Fig 7). The infectivity of the virus, analyzed by a transduction assay was not impaired by the process with similar transducing units per VG in harvest and final samples (data not shown).
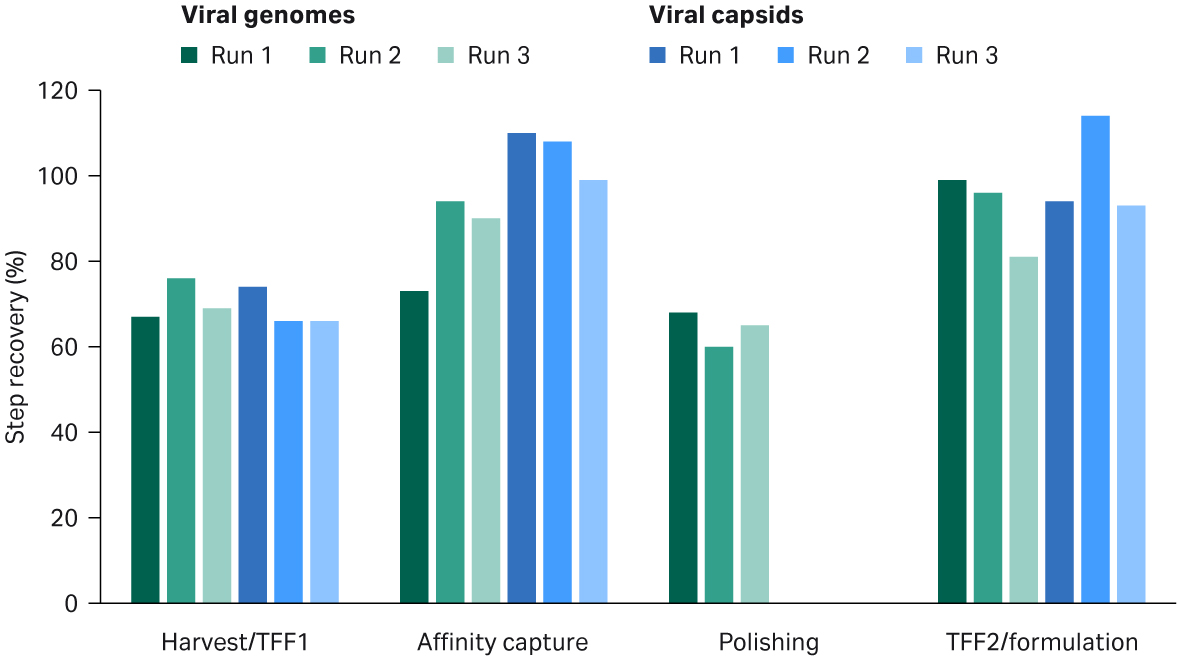
Fig 7. rAAV5 recoveries of viral genomes (green bars) and viral capsids (blue bars) for key process steps.
Impurity levels were assessed by HCP ELISA assay and hcDNA was measured by HEK293 qPCR. The levels of both HCP and hcDNA in the final product were below the regulatory limits according to the WHO guidelines (100 ng HCP/dose and 10 ng hcDNA/dose) calculated for a dose with 1 × 1011 VG (Fig 8 and Table 4). A dose of 1 × 1012 was also possible with impurity levels just below the regulatory limits. Residual nuclease was below the detection limit (data not shown) and Tween™ 20 was previously shown to be detected only in very low amounts after the first TFF step in a similar process. Samples from the different process steps were analyzed by SDS-PAGE and Western blot (Fig 9).
The concentration and purity level of the rAAV5 in the different process steps showed high HCP levels in the TFF material before affinity capture which was reduced to very low levels in the elution pool. In the rAAV5 Capto™ AVB eluate, two low MW protein bands could be seen that were not seen during process development. These proteins were removed in the following steps (Fig 9). Cryo-transmission electron microscopy (TEM) analysis was also performed to characterize the rAAV5 particles. The expected rAAV particle size of around 25 nm and a high sample purity can be seen in Figure 10. No aggregation of rAAV5 could be detected in the final samples from the three runs by analytical size exclusion chromatography with Superose™ 6 Increase (data not shown).
We have developed a scalable and GMP compatible start to finish rAAV5 production process including upstream production, downstream purification, and new analytic assays. We have shown three reproducible runs at 10 L scale production and purification with good VG productivity and enrichment of full capsids to above 60%, depending on the analysis method. The upstream production protocol was also applied to other serotypes (rAAV8 and rAAV9). Affinity capture with Capto™ AVB is compatible with AAV1, AAV2, AAV3, AAV6, and some other engineered capsids, however, the protocol may need to be adjusted for optimal performance.
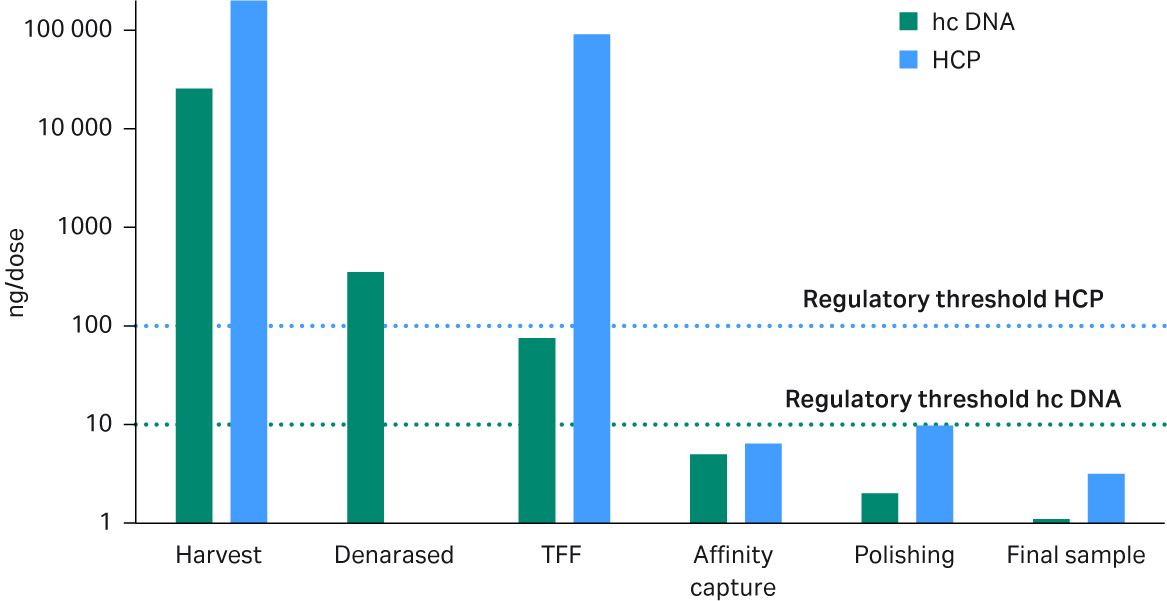
Fig 8. Analysis of HEK293T DNA (qPCR) and HCP (ELISA) shows that the regulatory limits of host cell impurities per dose (1 × 1011 viral genomes) were met after the affinity capture step.
Table 4. Analysis of final sample from the three process runs
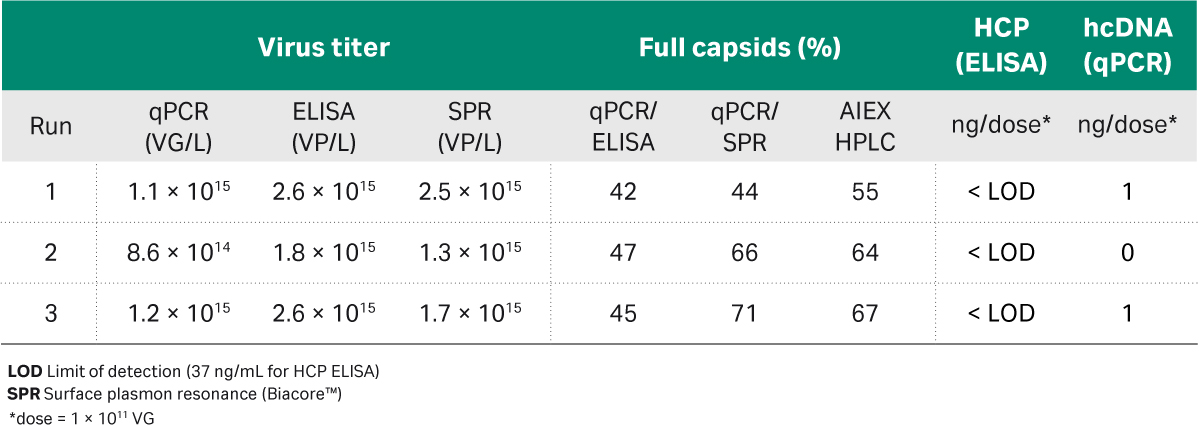
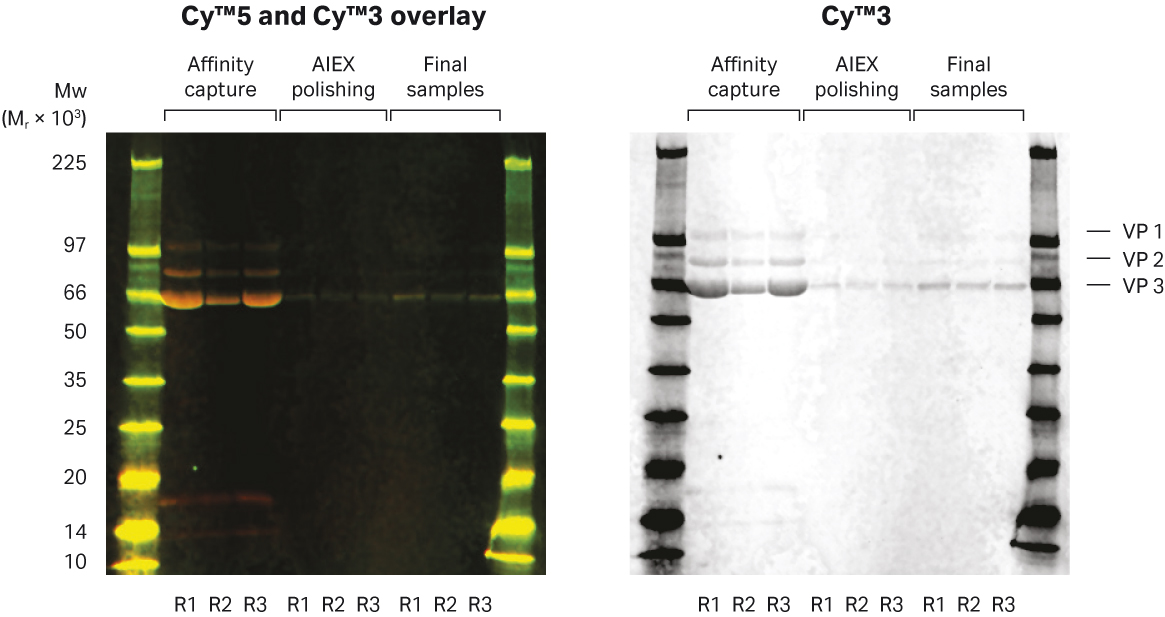
Fig 9. Analysis of process samples with multiplex fluorescence Western blot analysis showed HCP (Cy™5 prelabeled) and the presence of the three viral proteins (targeted with primary antibody and Cy™3-labeled secondary antibody).

Fig10. Cryo-TEM imaging of affinity-purified rAAV5 containing approximately 10% full capsids. Higher magnification shows a full and an empty capsid.
Conclusion
In conclusion, we have developed a scalable process for the production of rAAV using triple plasmid transfection of suspension cells in an animal-derived component-free medium. The cell culture process can be used for various serotypes with good productivity of viral genomes (approximately 1 × 1013 VG/L, and ≥ 10% full capsids). The purification process is based on filtration, efficient affinity capture, and polishing with > 3-fold full capsid enrichment. Both novel and established analytical methods were used for virus analysis, and results show that the material meets regulatory requirements concerning virus purity. The described process is compatible with GMP in single-use as well as with hard-piped process equipment.
Reference
- Werle, Amanda K., Powers, Thomas W., Zobel, James F., et al. Comparison of analytical techniques to quantitate the capsid content of adeno-associated viral vectors. Mol. Ther.: Methods & Clin. Dev. 2021;(23):254-262. DOI:https://doi.org/10.1016/j.omtm.2021.08.009
CY25645-19Apr22-WP
TR 29792295, 29794423, 29777055