The results of proteome analysis generates a complex mixture that relies on high throughput methods such as 2D gel electrophoresis and mass spectrometry (MS). To realize the potential of these advances in speed, sensitivity, and throughput of proteome analysis, sample preparation needs to catch up. As a result, simple processing of multiple samples has become the focus. Single-pot, solid phase, sample preparation (SP3) offers advances in sample prep for proteomics analysis allowing you to better understand the proteome’s complex makeup and to delve deeper in search of actionable biomarkers.
Introduction
Proteome analysis has historically been performed using 2D gel electrophoresis (2D GE). 2D gel electrophoresis generates separation maps based on isoelectric point and protein mass. Various iterations in separation have also been developed, such as, difference gel electrophoresis (DIGE) using Cy™ dyes to differentially label samples, that can then be run in the same separation.
Mass spectrometry (MS) has also been used to identify individual proteins following excision from a gel. HPLC and LC can be used for the separation and identification of low-molecular-weight proteins and peptides and can feed into a MS workflow. While 2D gel electrophoresis continues to be used it has been largely replaced by MS. Advancements in technology now allow thousands of proteins to be identified in a single run with sample sizes of only a few nanograms (Fig 1).
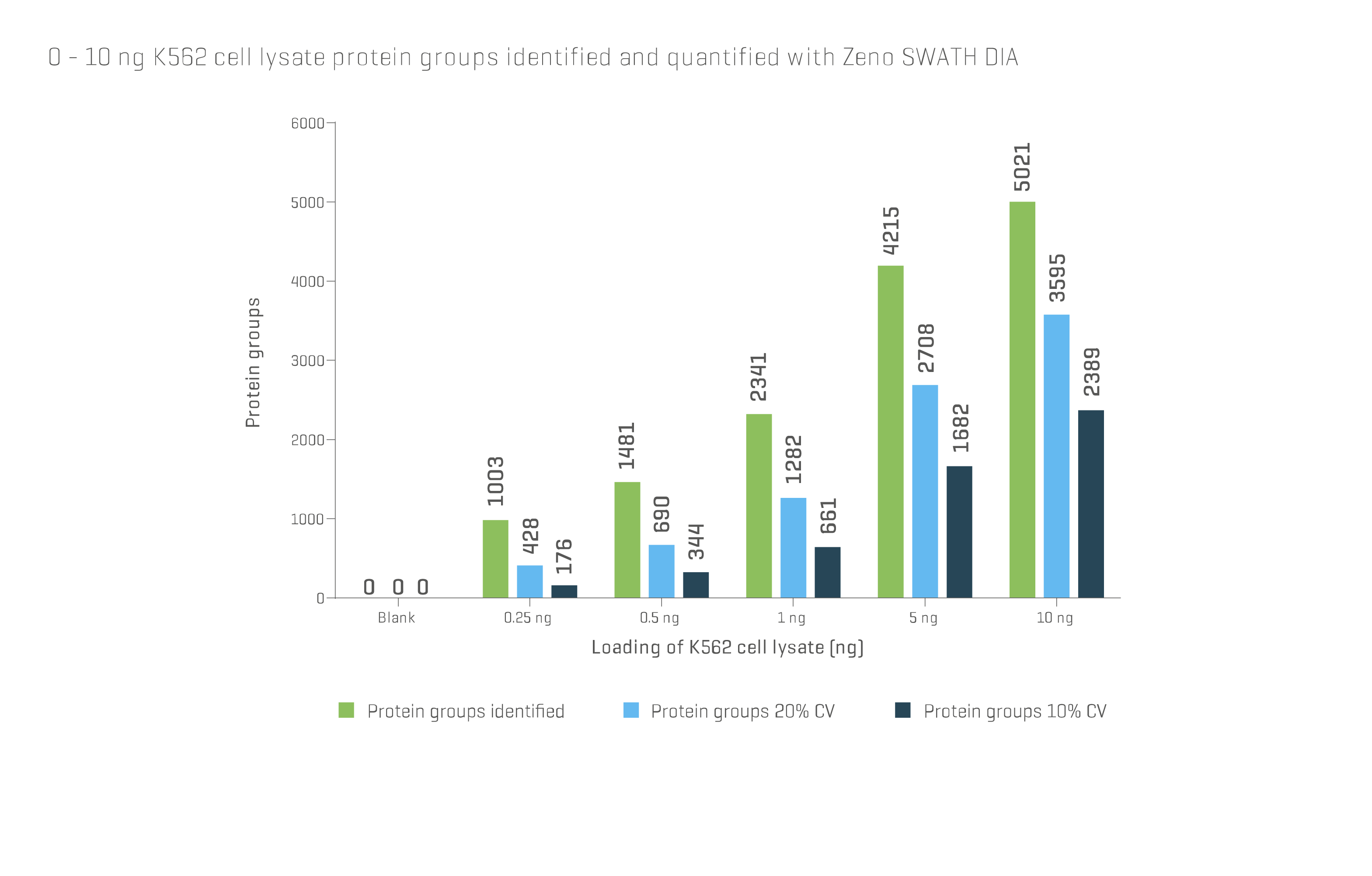
Fig 1. Data obtained from SCIEX® ZenoTOF® system using Zeno SWATH DIA shows the number of distinct protein groups identified and quantified by mass spectrometry can reach several thousand, even at sample loading amounts well below 10 ng. For further details, visit ZenoTOF 7600 system (sciex.com)
Simplified workflows
To realize the potential of these advances in speed, sensitivity, and throughput of proteome analysis, sample preparation needs to catch up. As a result, simple processing of multiple samples has become the focus. In recent years, paramagnetic particles have been used to capture the total proteome as an alternative to established methods, such as, filter-aided sample preparation (FASP) (1).
This method, commonly referred to as single-pot, solid phase, sample preparation (SP3), employs a mixture of E3 and E7 carboxyl functionalized Sera-Mag™ particles from Cytiva. SP3 was introduced by Hughes et al. and is gaining popularity due to the speed and ease of use offered (2).
Several studies have compared SP3 with existing methods, for example, Supasri et al. compared SP3 with FASP for bottom-up shotgun proteotyping in Symbiodiniaceae (2). They concluded that the SP3 protocol provided better peptide detection data, with the single-pot operation preventing sample loss during processing, especially important for smaller inputs.
In a separate study, Mikulášek et al. also benchmarked SP3 against the established FASP in a plant proteome bottom-up analysis (4). They concluded that SP3 offered better flexibility in terms of input range from 0.1 to 100 µg without adjustment, providing superior protein identification. Both studies highlighted the ease of use and significant time/cost savings in addition to the improved performance offered by SP3.
Learn more about Reproducible protein and peptide cleanup for mass spectrometry
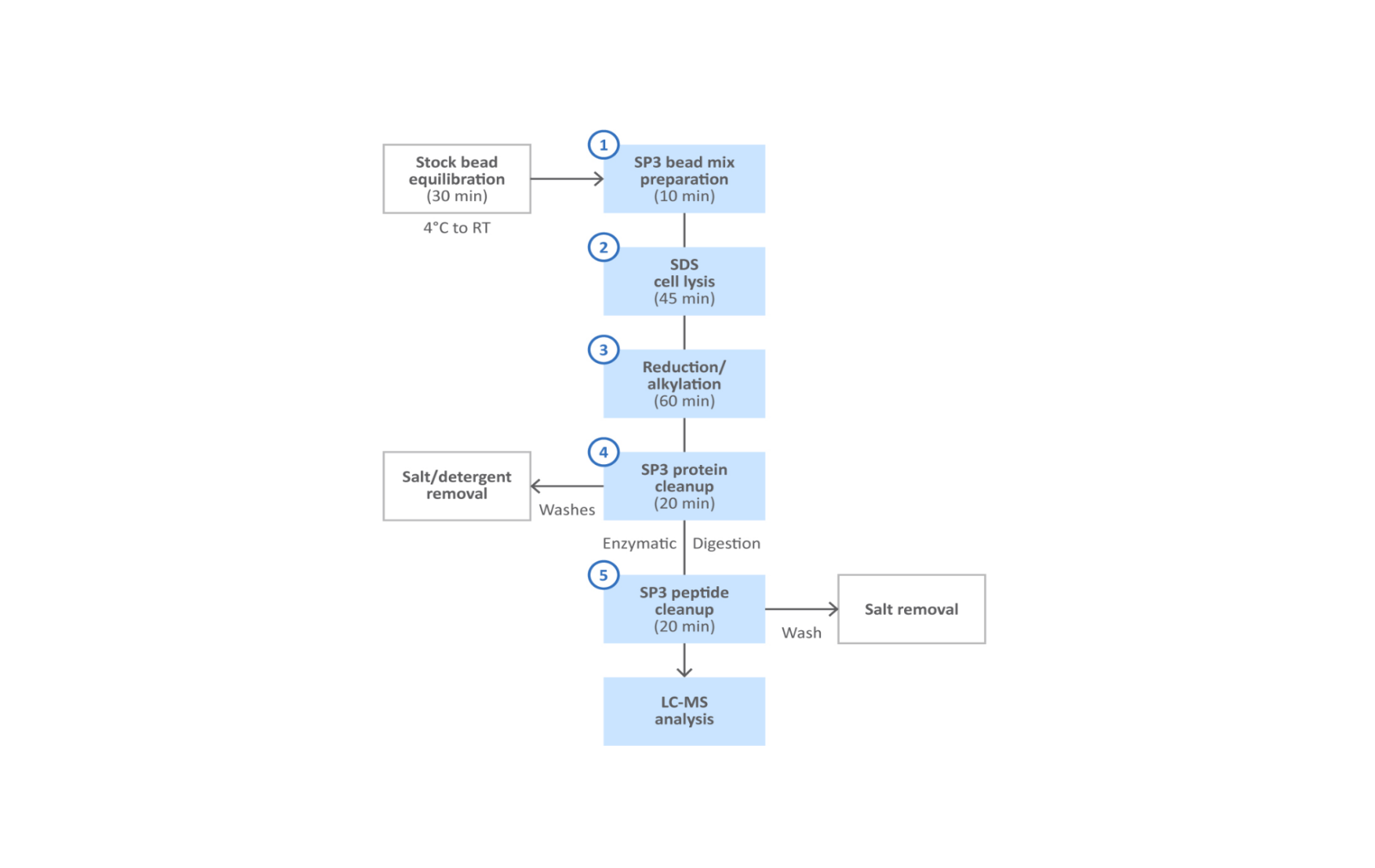
Fig 2. SP3 protein and peptide cleanup workflow.
A simplified workflow for SP3 is shown in Figure 2. With this method, the characteristic surface properties of the Sera-Mag™ magnetic particles enable effective and unbiased capture of total protein fraction (Fig 3). Following lysis, the protein fraction is bound to the beads in presence of organic solvents such as ethanol or acetonitrile. The magnetic particles are washed to remove detergents, salts, and other contaminants that remain in the supernatant. Captured proteins are treated with protease to digest into peptides, that accumulate in the supernatant. The peptides are then captured with fresh magnetic beads prior to final elution.
Request a sample of Sera-Mag™ carboxylate-modified magnetic beads
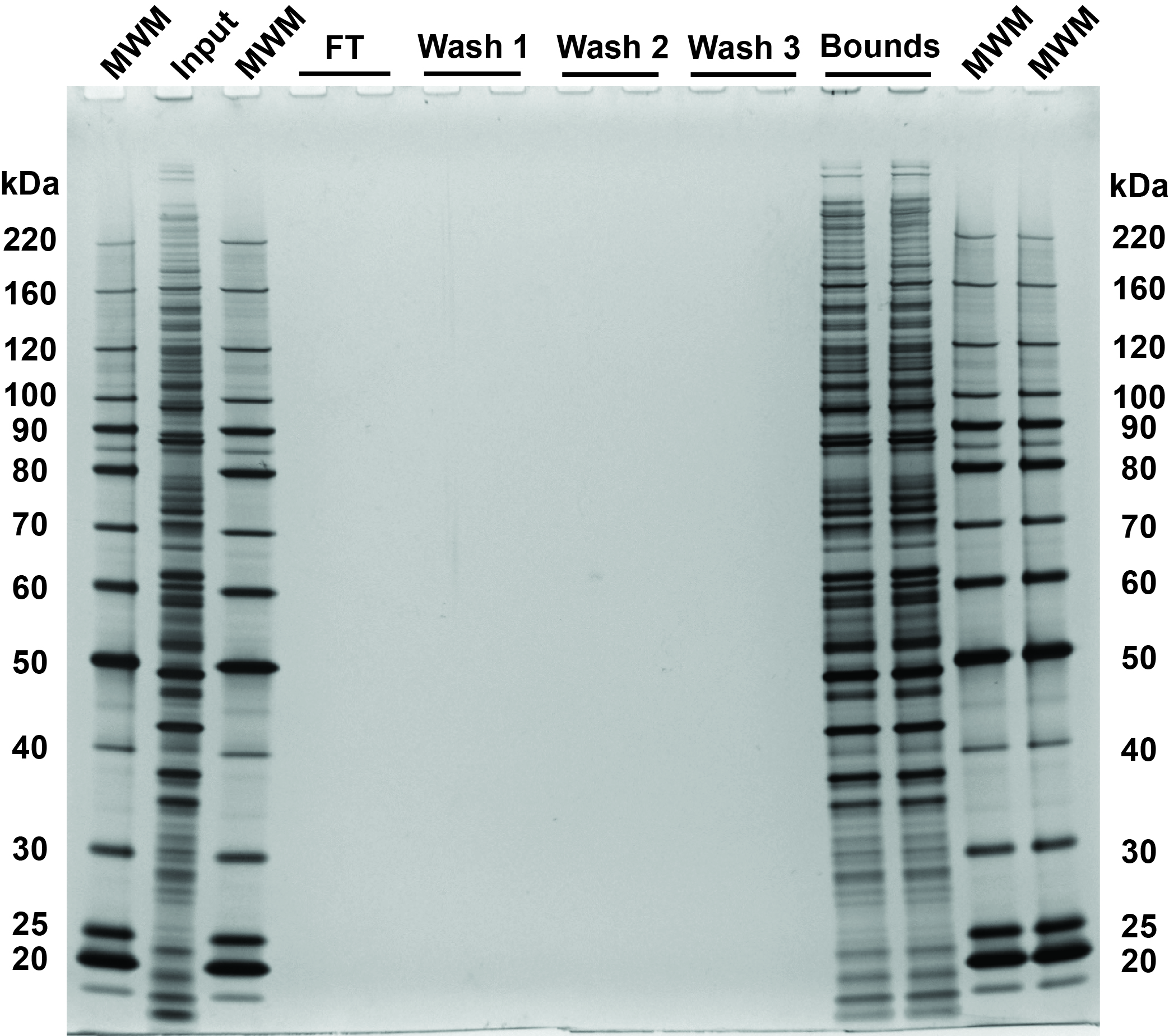
Fig 3. SDS-PAGE and silver stain visualization of 10 µg replicate SP3 protein cleanups. Comparison of input and bound fractions illustrates effectiveness of SP3 protein-level protocol. HeLa S3 cells were lysed in SDS, treated with benzonase, reduced and alkylated followed by SP3 cleanup. 0.5 µg of each fraction loaded on the gel.
Applications
Here we look at a few examples where SP3 is being utilized to gain insights:
Inflammatory response in primary sclerosing cholangitis
Kan et al. investigated the bile proteome identifying a total of 7889 proteins, significantly more than previous studies (5). Many of the proteins were related to proliferation and activation of inflammatory response which was specifically up-regulated in samples from patients suffering from primary sclerosing cholangitis. Analysis was carried out on 20 µg of protein per sample, isolated, and cleaned up using SP3. Resulting peptides were directly injected and run by DIA-MS. Following data analysis, several potential biomarkers such as interleukin-8 and annexin A1 (ANXA1) were identified for further study.
Refined characterization of chronic lymphocytic leukemia using proteomics
Chronic lymphocytic leukemia (CLL) is the most common type of Leukemia in western countries. Herbst et al. took a proteogenomic approach, characterizing the proteome and transcriptome alongside genetic and ex vivo drug profiling to explore differences in therapy response and progress beyond current diagnostics approaches (6). For the proteome analysis, they used SP3 to isolate the proteins, then following digestion, separated peptides using high resolution isoelectric focusing (HiRIEF) prior to MS analysis.
From six associated proteomic groups, the investigators found one group not detectable at the transcriptome level. (ASB-CLL), this group was characterized by accelerated disease progression, altered spliceosome, and low B cell signaling. This multi-omics analysis refines the classification of CLL and highlights the potential of proteomics to improve cancer patient stratification and personalized treatments beyond genetic and transcriptomic profiling.
Proteomics as a tool to explore heterogeneity in breast cancer
Asleh et al. performed comprehensive proteome profiling of 300 archival breast cancer specimens dating back as far as 1986 to investigate potential biomarkers and therapeutic targets (7). RNA-based gene signatures typically used to identify intrinsic breast cancer subtypes, do not always resolve the extensive heterogeneity that characterizes many breast cancers and so proteomic analysis is being explored to further characterize beyond currently defined subtypes.
With sample availability limited for such analyses, the investigators optimized SP3 to work with small amounts of FFPE specimens and were able to quantify over 9000 proteins with more than 4000 quantified across every sample. The study discovered separate proteome groups associated with distinct clinical outcomes and immune responses that fall within the same subtype classifications under RNA-based PAM50 currently. The investigators identified several protein candidates for in-depth analysis and provided a method that demonstrates global proteomic analysis on standard FFPE specimens in a reliable and clinically applicable manner.
SP3 offers new insights into biomolecular archaeology
SP3 has also found favor in the field of biomolecular archaeology. For example, Palmer et al., used the method to profile the proteome of human dental calculus (8). Here, the ability to maximize proteome recovery in ancient samples, available only in minute quantities of precious materials is of paramount importance. The team detected up to twice as many unique peptides and matched proteins compared to existing methods, including bacterial and dietary proteins.
In a similar archaeological study, Solazzo and Niepold, looked at methods to prepare hair and skin proteins from archaeological fur and leather at an early medieval burial ground dating to 560 CE (9). They optimized their SP3 protocol to successfully identify fur and leather samples to species, that was not possible with existing methods.
Summary
It is clear that mass spectrometry in proteome research has taken huge strides forward. To take advantage of these speed and resolution capabilities, sample preparation methods, such as SP3, which are simple, reliable, and sensitive are needed. In conclusion, we have shown that:
- SP3 using Sera-Mag™ carboxylated paramagnetic beads has proven to be a simple and robust methodology for unbiased capture of total proteins from complex matrices.
- SP3 is well suited to a wide range of sample types, from human and animal tissues to more difficult samples such as plant tissue, archaeological specimens, and even FFPE clinical samples.
- The single-pot method is also scalable, offering sensitivity, especially with smaller samples and especially difficult samples, enabling detection of more unique peptides and associated protein groups.
Related content
Application Note: Reproducible protein and peptide cleanup for mass spectrometry
A spotlight on magnetic beads
The scientist’s guide to magnetic beads
Magbeads 101: A guide to choosing and using magnetic beads
Test your knowledge of magnetic beads
References
- Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359-62.
- Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, Krijgsveld J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol. 2014;757.
- Supasri KM, Kumar M, Mathew MJ, et al. Evaluation of Filter, Paramagnetic, and STAGETips Aided Workflows for Proteome Profiling of Symbiodiniaceae Dinoflagellate. Processes. 2021;9(6):983.
- Mikulášek K, Konečná H, Potěšil D, Holánková R, Havliš J, Zdráhal Z. SP3 Protocol for Proteomic Plant Sample Preparation Prior to LC-MS/MS. Front Plant Sci. 2021;12(635550).
- Kan M, Chiba T, Konno R, et al. Bile proteome analysis by high-precision mass spectrometry to examine novel biomarkers of primary sclerosing cholangitis. J Hepatobiliary Pancreat Sci. 2022 Dec 17.
- Herbst SA, Vesterlund M, Helmboldt AJ, ,em>et al. Proteogenomics refines the molecular classification of chronic lymphocytic leukemia. Nat Commun. 2022;13(6226).
- Asleh K, Negri GL, Spencer MS, ,em>et al. Proteomic analysis of archival breast cancer clinical specimens identifies biological subtypes with distinct survival outcomes. Nat Commun. 2022;13(896).
- Palmer KS, Makarewicz CA, Tishkin AA, Tur SS, et al. Comparing the Use of Magnetic Beads with Ultrafiltration for Ancient Dental Calculus Proteomics. J. Proteome Res. 2021;20(3):1689-1704.
- Solazzo C, Niepold T. A simplified sample preparation for hair and skin proteins towards the application of archaeological fur and leather. Journal of Proteomics. 2023;274(104821)