Introduction
Lentiviral vectors (LVs) are used in cell and gene therapy to deliver therapeutic genetic material to target cells and tissues. Traditionally, LVs have been manufactured in adherent cell cultures using transient transfection to deliver viral packaging genes that are encoded on three or four plasmids. Process scalability, lot-to-lot consistency, and the cost and supply of high-quality plasmid are some of the constraints that have made manufacturing of LVs challenging.
Adapting stable LV producer cells to suspension culturing in serum-free medium may resolve some of the issues related to scalability, consistency, and the supply chain for large-scale LV production. HEK293SF-LVP-CMVGFPq-92 (clone 92) cells, developed by the National Research Council of Canada (NRC), are a model stable producer cell line encoding a third generation LV-GFP, conditionally self-inactivated. The NRC’s clone 92 cells are induced for LV production by addition of small molecules (cumate and doxycycline), which activate transcription of the viral rev and vsv-g (glycoprotein envelope) genes. The cells are stable in culture without selection and can maintain LV production over many passages.
The process of using stable, inducible producer cells for LV production consists of a preinduction cell expansion phase, induction of LV production by addition of chemical inducers, and harvesting of vector from the culture at an appropriate point (or points) during the production window. However, several culturing strategies (e.g., batch, fed-batch, and perfusion feeding) can be used to culture the model clone 92 cells and produce LV-GFP (as described in Manceur et al. 2017; 1). The same culturing strategies could be applied for other producer cells that may package more clinically relevant transgenes.
The choice of a suitable approach will depend on specific process constraints (e.g., cost, scale, available resources, etc.) and needs (e.g., yield), which must be determined ahead of process development activities and are used to design and define a process that is fit for its purpose. Here, we demonstrate a high-yield LV production process, using animal-derived component-free (ADCF) media and reagents, where multiple or continuous vector harvests could be obtained from a single batch.
See detailed materials and methods.
Results and discussion
Previously, clone 92 cells were used to develop a linearly scalable LV production process, where a single harvest of vector was collected at the end of the batch culture. In this study, the goal was to develop a higher yield process that allowed regular or continuous vector harvesting in conjunction with replacement of harvested medium with fresh medium.
Initial development work was performed in small scale, using cells cultured in shake flasks, to identify a suitable medium and culturing strategy to scale up to a 1 L stirred-tank reactor (STR) culture. viral production in particular was seven times higher in a 1 L STR culture, using perfusion to harvest virus and replace spent medium with fresh medium.
Small-scale process with fortified batch culture and daily medium replacement
The multiple-harvest LV production process tested at small scale (in shake flasks) consisted of a preinduction expansion of clone 92 cells in a batch culture, followed by induction for vector production (by additions of doxycycline and cumate at appropriate concentrations), and vector harvesting and medium replacement at regular 24 h intervals for the next five days after the induction point. This enabled an approximate perfusion rate of one reactor volume per day, which would be used in the scale-up of the process. To better mimic the gradual medium exchange in perfusion-based 1 L STR and not expose cells to fully fresh medium all at once, only 50% of the spent medium was harvested and replaced with fresh medium each day.
In the previous study viable cell density (VCD) at induction was linearly correlated to volumetric LV production, but only when cells were induced in the exponential phase of their growth. Therefore, prior to executing the small-scale process, supplementation of the culture medium was interrogated as a strategy to increase the VCD at induction and consequently volumetric titers of the vector (transducing units/mL = TU/mL).
The fully chemically defined HyClone HyCell TransFx-H medium, supplemented with 4 mM L-glutamine and 0.1% poloxamer 188, was initially used to culture cells in suspension. When inoculated at a VCD of 2.5 × 105 cells/mL, clone 92 cells reached a maximum VCD of ~ 5 × 106 cells/mL after seven days in culture (Fig 1). However, cell viability and doubling time decreased after four days in the culture. Therefore, HyClone Cell Boost 5 (CB5), a fully chemically defined cell culture supplement containing lipids, amino acids, vitamins, and growth factors, was added to the culture medium at a concentration of 3.5 g/L (using a 35 g/L stock solution added as a 10% volume/volume medium supplement) to grow cells to a higher VCD. Addition of CB5 to the culture medium allowed cells to reach a maximum VCD of ~ 1 × 107 cells/mL with viability > 90% in batch culture (Fig 1).
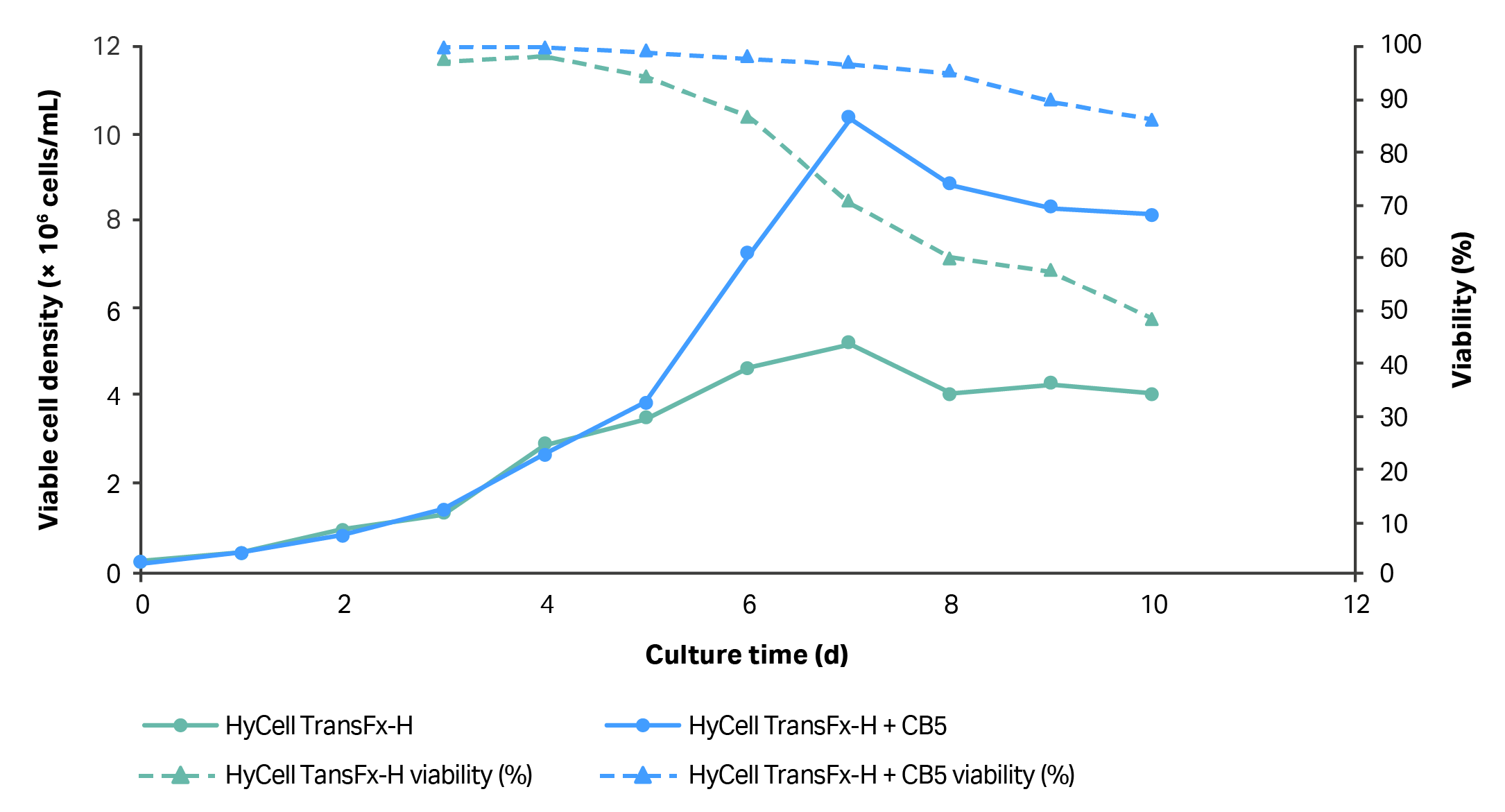
Fig 1. Growth of clone 92 cells in HyCell TransFx-H medium and supplemented with Cell Boost 5 (3.5 g/L). N=2 (each data point is average of two readings from one process).
For LV production, clone 92 cells were inoculated at a VCD of 2.5–4.0 × 105 cells/mL in 100 mL of HyCell TransFx-H medium supplemented with CB5 (3.5 g/L), 4 mM L-glutamine, and 0.1% poloxamer 188. The cells reached a density 5.5 × 106 cells/mL after five days in culture, which was chosen as the induction point based on data generated in Figure 1 and the requirement for cells to be in the mid-exponential phase of growth for best specific productivity. Four discrete 20 mL aliquots of this culture were induced to produce LV (by direct additions of concentrated inducers to culture medium). For each cell suspension, 50% of the culture medium was exchanged with fresh medium containing inducers, with or without CB5 supplementation, at every 24 h following induction.
Production of vector was expected to result in decreased viability of cells due to the expression of cytotoxic proteins (viral protease and VSV-G) during the process. Interestingly, clone 92 cells that were continually cultured in medium supplemented with CB5 showed higher VCD and viability relative to non-CB5 supplemented cells, starting at two days post-induction and continuing to the end of the process (Fig 2). At the end of the ten-day expansion and induction process, the cells cultured in medium, which was continually supplemented with CB5, were on average 86% viable compared to 37% viable when replacement medium did not contain CB5. Similarly, the VCD at the end of ten days was more than 6-fold greater in cultures when replacement medium contained CB5.
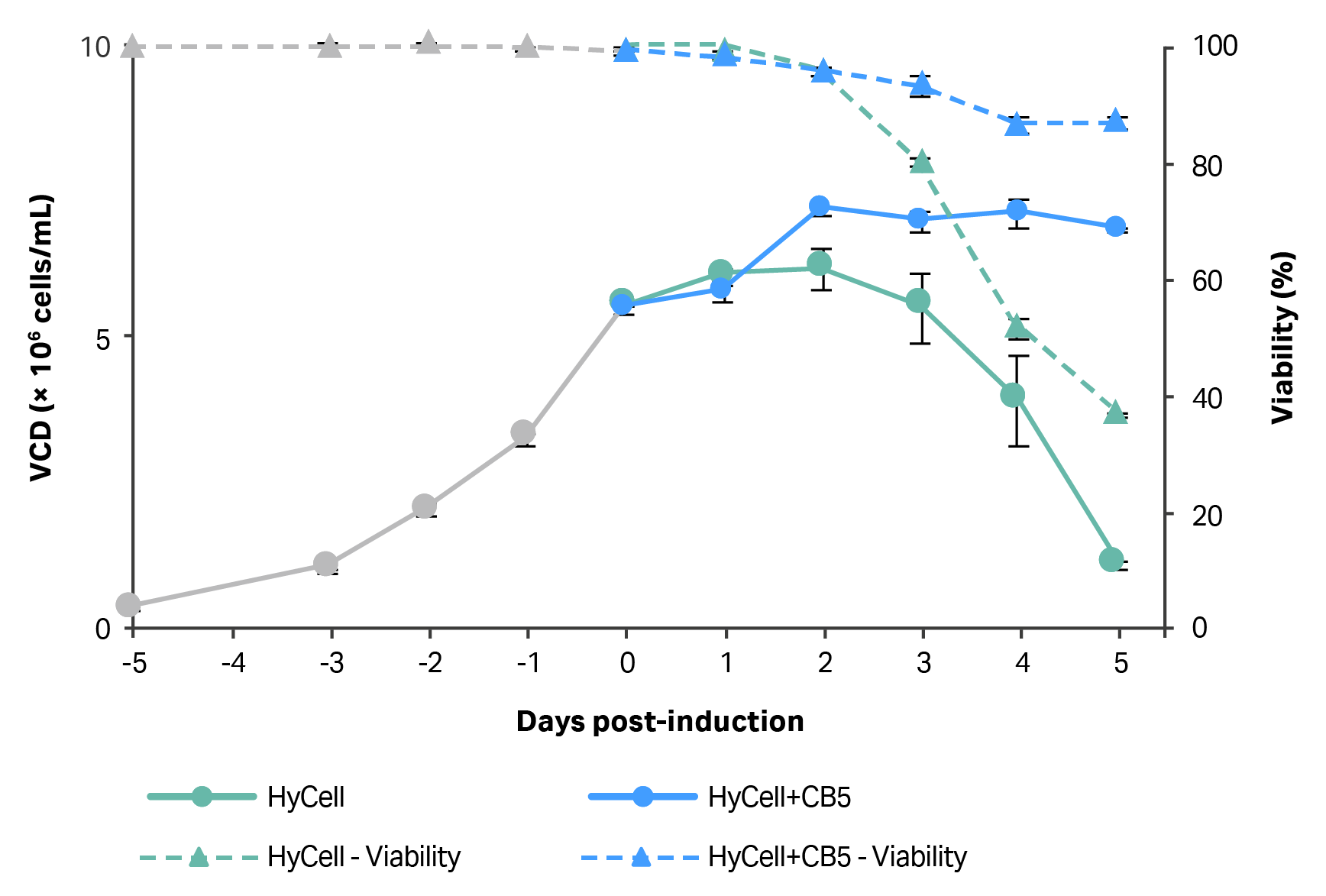
Fig 2. VCD and cell viability of clone 92 cells prior to and after induction of LV production in 20 mL cultures. Half of medium was replaced with HyCell TransFx-H or HyCell TransFx-H supplemented with CB5 (3.5 g/L), starting at 24 h after induction and every 24 h thereafter. N=2 (each data point is average of two readings from one process).
In terms of LV production, similar volumetric functional titers, ~ 4 × 107 TU/mL, were observed for harvests made from all cultures at two- and three-days post-induction, regardless of CB5 supplementation in the replacement medium (Fig 3). However, higher functional titers were observed in harvests made from cultures where replacement medium contained CB5 than when it did not at both four- and five-days post-induction; the difference was more pronounced for the latter. However, the difference in titers on both days was not proportional to the difference in VCDs observed between the two conditions at corresponding time points. The differences in VCDs between the two feeding strategies were larger proportionally than the differences in titers, suggesting that cell-specific productivity may have been lower in the condition where CB5 was included in the replacement medium.
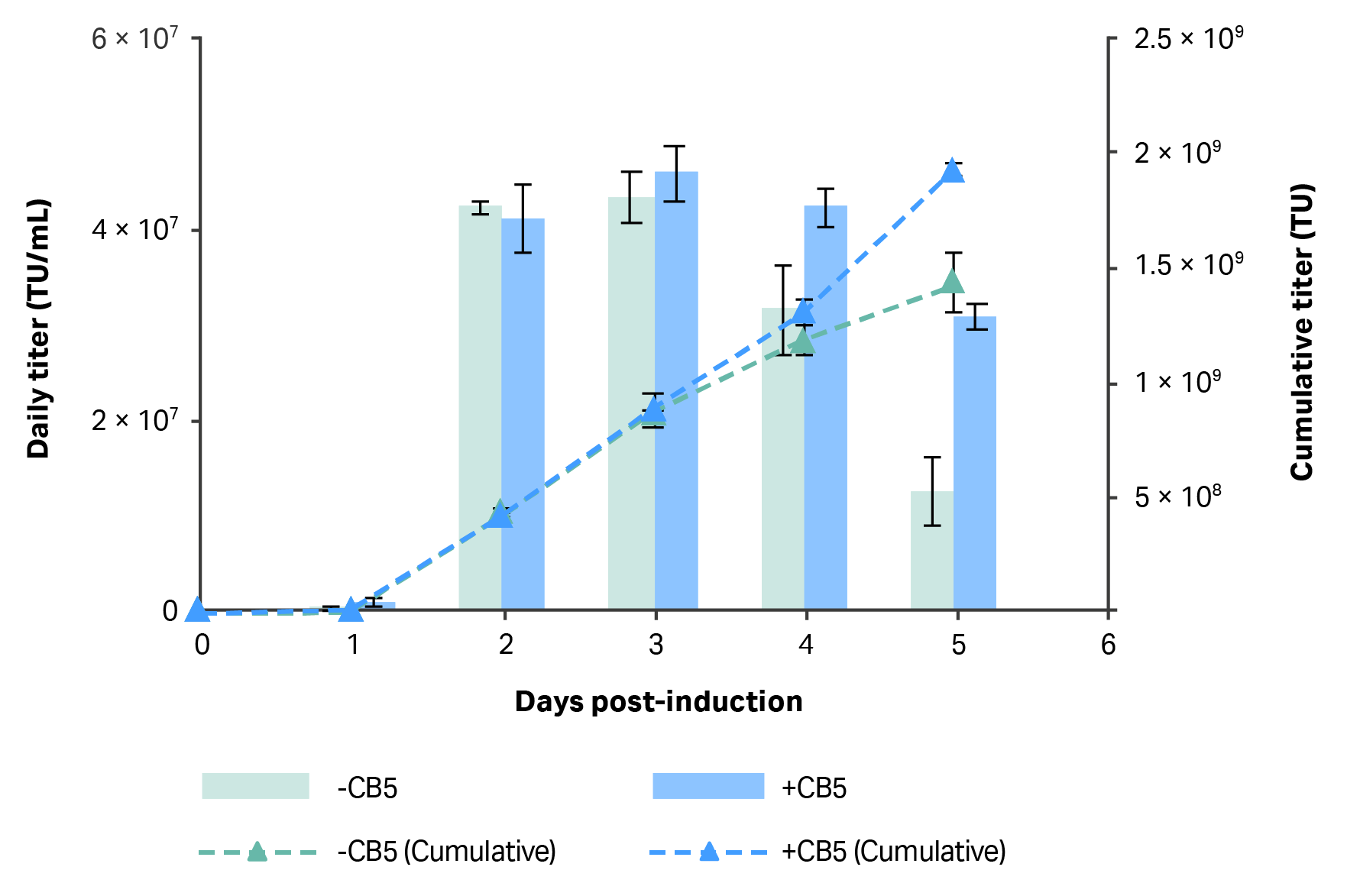
Fig 3. Daily volumetric vector titer (TU/mL) in 20 mL cultures where 50% of medium was replaced daily with fresh HyCell TransFx-H or HyCell TransFx-H supplemented with CB5 (3.5 g/L) starting at 24 h after induction. N=2, two technical samples to measure infectious titer (with three dilution replicates for each sample).
CB5 was included in the replacement medium for scale-up to a 1 L STR culture despite its apparent negative effects on cell specific productivity. The main reasons were CB5 had a positive impact on volumetric productivity (TU/mL) and prolonged the production window past the third day following induction, resulting in greater overall yield than when HyCell TransFx-H medium was used alone (Fig 3).
Translation to 1 L STR with perfusion-enabled approach for LV production
To translate the small scale, shake flask process to a 1 L STR culture, the use of centrifugation to enable regular medium collection and replenishment during LV production was replaced by perfusion feeding. This was done to simplify the workflow and minimize manipulation of the culture during LV production, to allow for continuous vector harvesting if desired, and for ease of scalability at volumes exceeding 1 L of culture. Other features of thisprocess, including the induction VCD and the production medium remained unchanged from the small-scale process. However, some differences in performance were expected between the two scales, because the changes in the medium replacement strategy were considered impactful on vector accumulation kinetics and the metabolic state of the producer cells.
The process at 1 L scale consisted of the following steps:
- A 200 mL culture of clone 92 cells was seeded into 800 mL of HyCell TransFx-H medium, supplemented with CB5 (3.5 g/L), 4 mM L-glutamine, and 0.1% poloxamer 188 to achieve a starting VCD of 2.5–4.0 × 105 cells/mL.
- Cells were batch cultured until the day that VCD reached ≥ 5 × 106 cells/mL.
- Cells were then induced to produce virus by addition of concentrated chemical inducers to culture medium and perfused at a rate of one reactor volume per day with fresh medium containing the same chemical inducers for five to seven days (Fig 4).
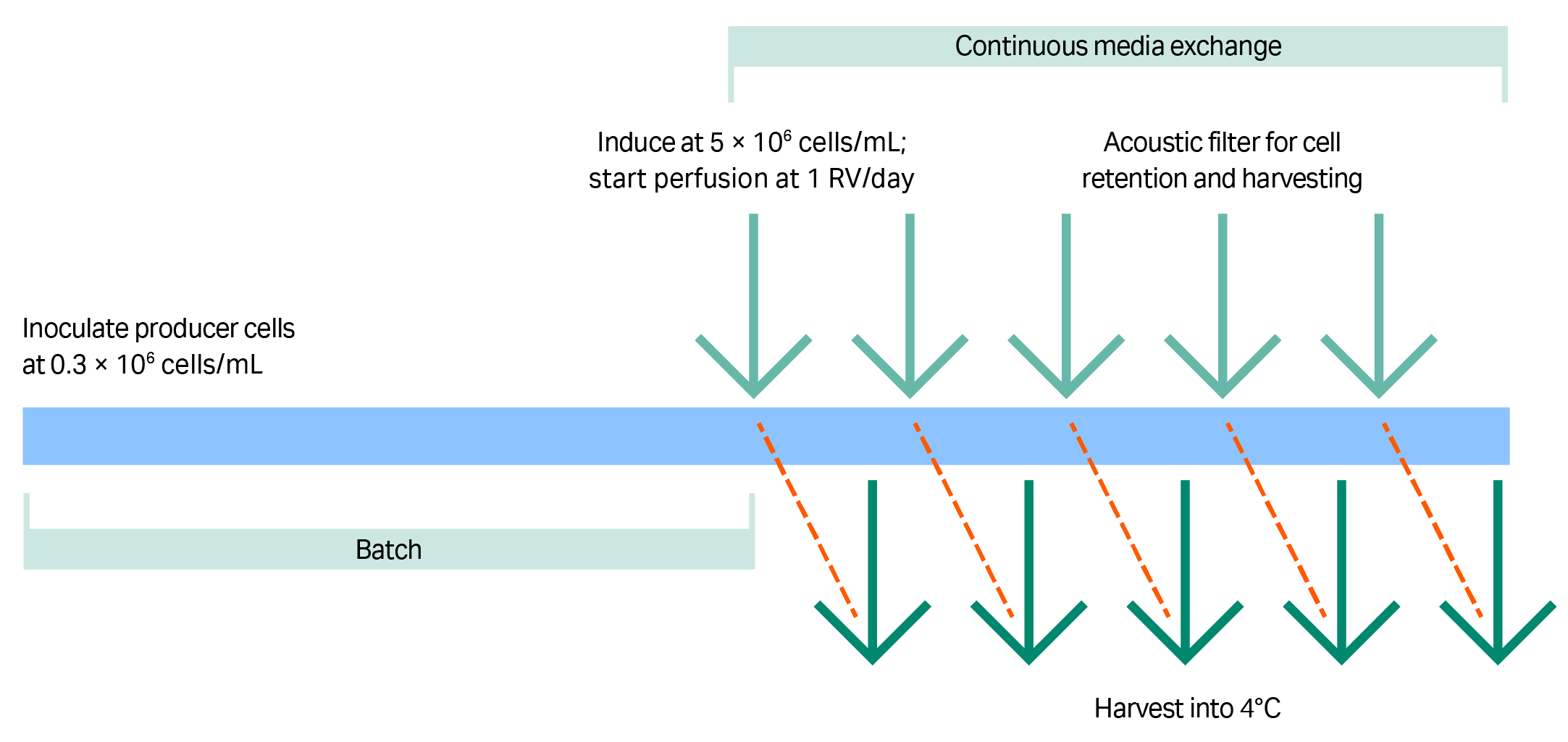
Fig 4. Daily volumetric vector titer (TU/mL) in 20 mL cultures where 50% of medium was replaced daily with fresh HyCell TransFx-H or HyCell TransFx-H supplemented with CB5 (3.5 g/L) starting at 24 h after induction. N=2, two technical samples to measure infectious titer (with three dilution replicates for each sample).
Filter type is critical for continuous harvesting of LV production strategies, because many perfusion filters have pores that are too small to accommodate LV passage (as we have observed in our laboratory). In this study, an acoustic filter was used for cell retention during perfusion and to allow LV to pass into the harvest (Fig 5). The spent medium containing the vectors was collected at 4°C in daily harvests that totaled one reactor volume per day.
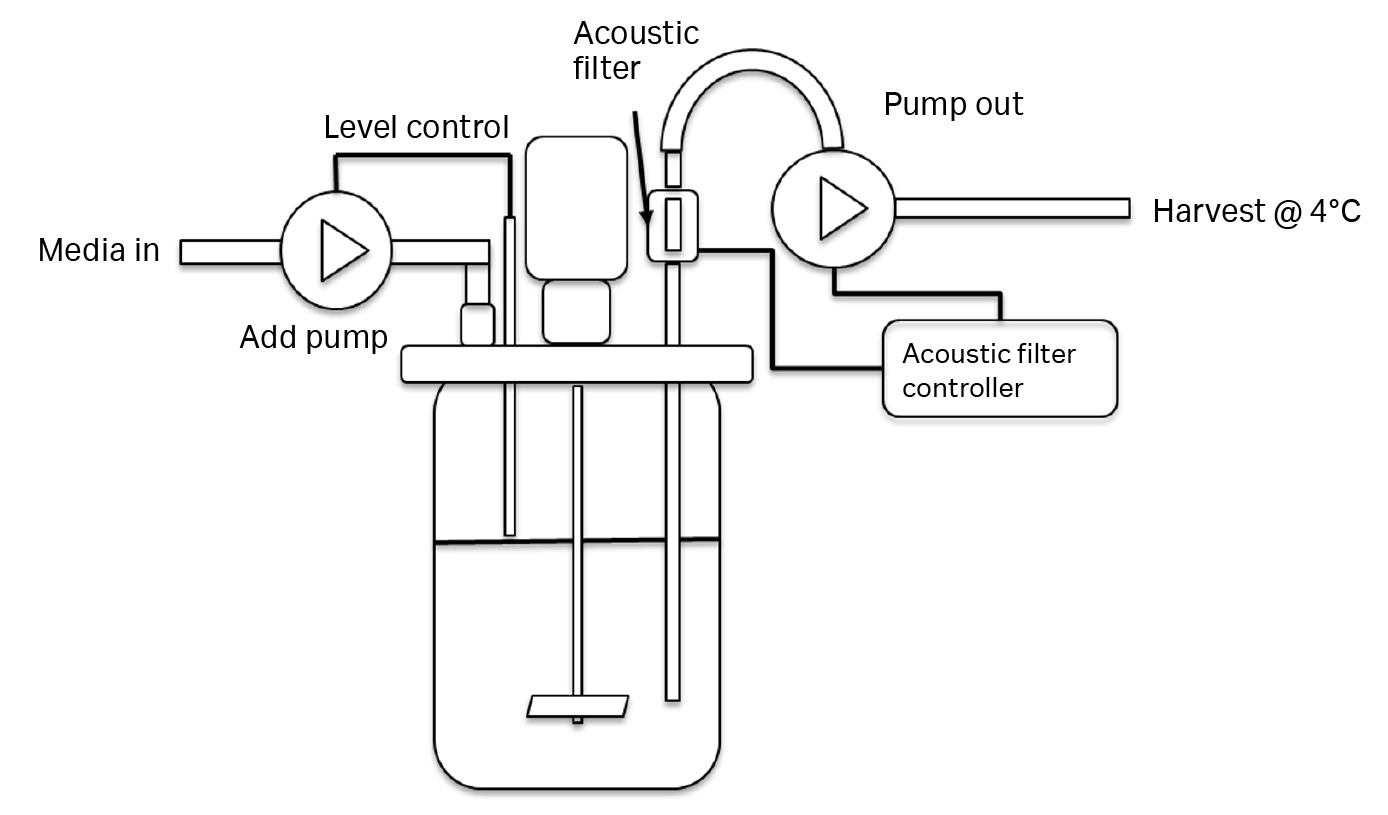
Fig 5. STR set-up detailing the perfusion strategy. The level sensor controls the fresh medium addition via ‘add pump’. The ‘pump out’ is continuously operating in a forward pump/reverse pump cycle (to flush back trapped cells) with a net pump-out volume of one reactor volume per day.
Seven runs were performed in total. Five runs followed the strategy as detailed in Figure 4, where perfusion with fresh medium started at time of induction. In run 3, perfusion with fresh medium was started one day after induction and in run 7, perfusion was started one day prior to induction. These changes were made to test the effect of the timing of starting perfusion on LV production kinetics and yield.
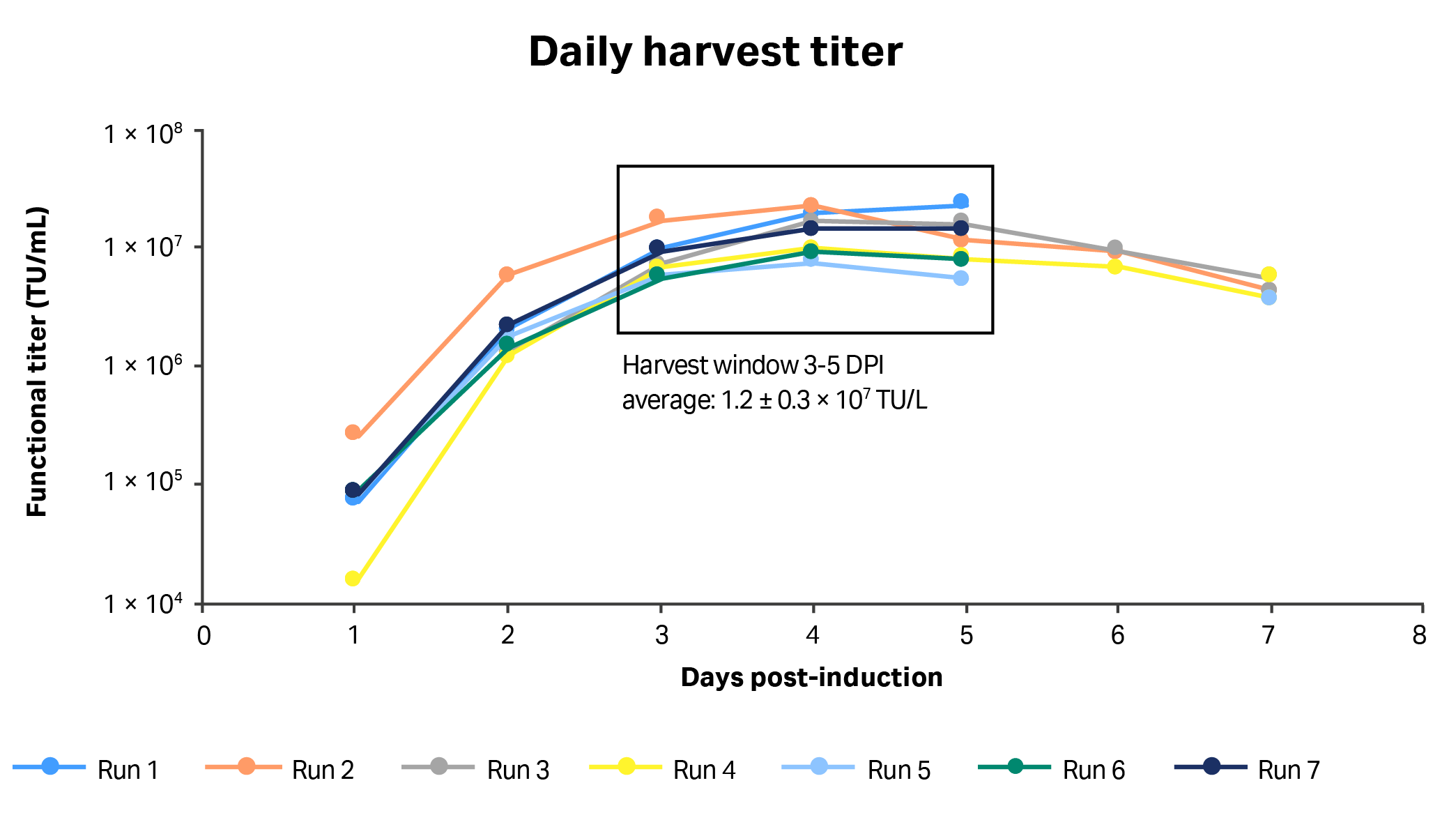
Fig 6. Daily volumetric vector titers (TU/mL) from each run in 1 L STR cultures using clone 92 cells and a perfusion-enabled process. Harvest windows: 3 to 5 d post-induction (DPI).
In all seven runs, the highest functional titers were harvested at three, four, and five DPI, with an average functional vector titer of 1.2 ± 0.3 × 1010 TU/L across this period. No apparent differences in yield and LV production kinetics were observed between runs 3 and 7 where perfusion was started one day after and one day before induction, respectively. In runs 2, 3, and 4, where LV production was extended to seven days post-induction, functional vector titer was significantly lower in harvests collected seven days after induction than in those collected after six days (paired T-test, p < 0.05). In all runs, the cell viability remained greater than 80% throughout the process and VCD increased after induction of LV production (data not shown).
The eventual decrease in vector production, without a corresponding decrease in VCD, indicated that specific productivity of clone 92 cells decreased after peaking three to five days after induction point under the tested conditions. The decrease in cell-specific LV productivity over time was expected based on data generated in small-scale runs. However, the kinetics of LV production in the 1 L STR culture were somewhat different than what was observed in a small scale, where LV titers peaked at two, three, and four days after induction. Also, the observed average titers during the peak three-day production periods were 3.6-fold lower in the 1 L STR than in the small-scale cultures (1.2 × 107 TU/mL compared to 4.3 × 107 TU/mL, respectively).
Some of the phenomena can be explained by the difference in harvesting method between the two scales. In terms of LV concentration in the harvest, the 1 L STR cultures were disadvantaged relative to the small-scale cultures, because the harvests collected by perfusion were being diluted in an equal volume of fresh medium as they were collected (i.e., fresh medium entering the reactor was also nonselectively being evacuated from the reactor by the ‘pump out’, a feature of all perfusion-fed cultures). Therefore, the volumetric titers were expected to be around half of those observed at small scale, had all else been equal. Also, similar cumulative LV yields were expected relative to culture volumes, since only 50% of the culture volume was harvested daily in the small-scale process.
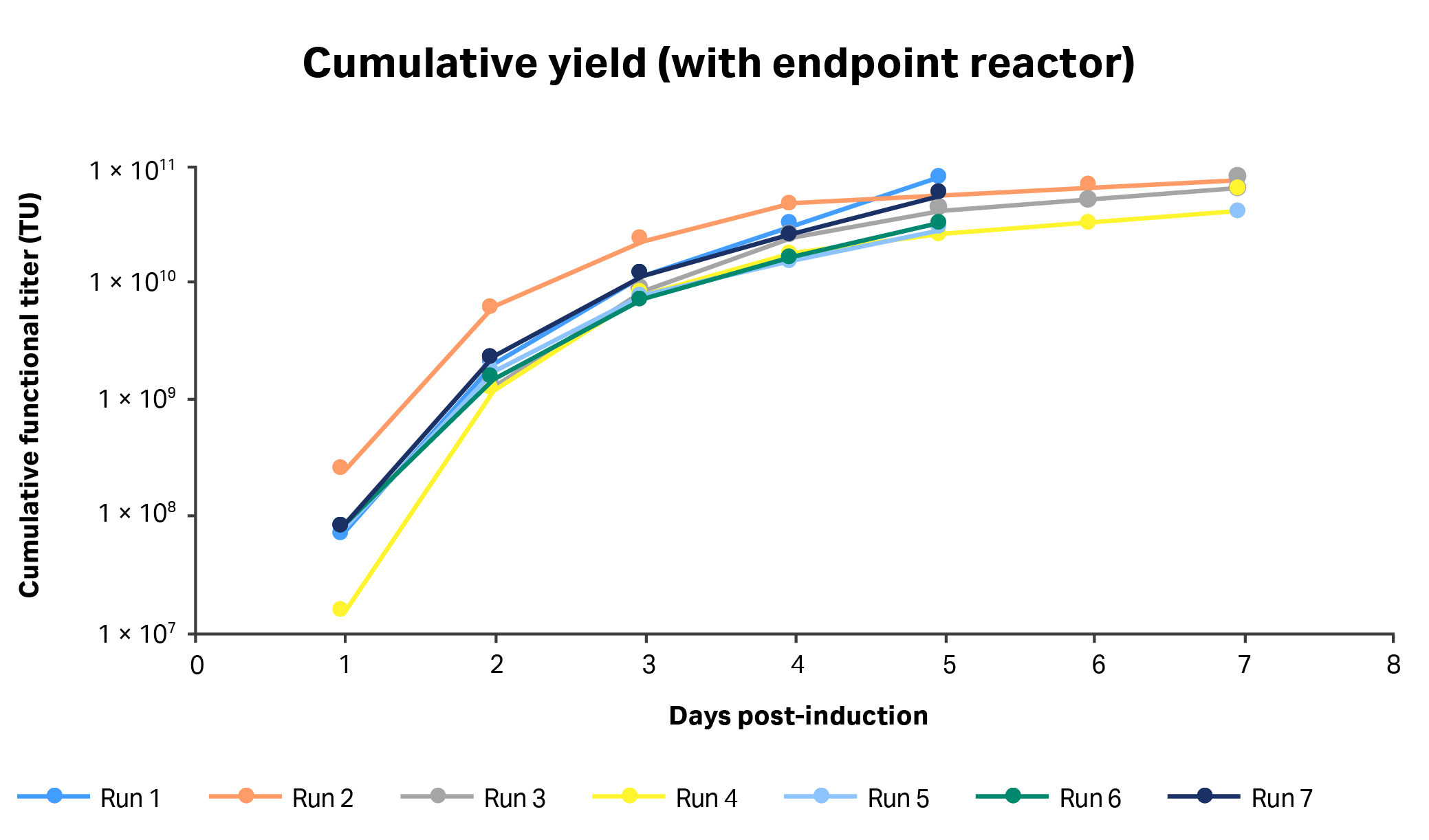
Fig 7. Daily volumetric vector titers (TU/mL) from each 1 L STR culture run using clone 92 cells and a perfusion-enabled process.
The cumulative yields of the 1 L STR processes after five days of induction were on average 5.2 ± 1.9 × 1010 TU, which included all the collected harvests and the contents of the reactor on the final day of culture (average of 1.4 ± 0.6 × 1010 TU) (Fig 7 and 8). If titers observed in the small-scale process were extrapolated to a 1 L culture, the cumulative yield, including the shake-flask contents on the last day, would be 9.6 × 1010 TU, or 1.9-fold greater than for the 1 L STR process. However, unlike in the small-scale process where vector had been harvested at discrete points in the process, the daily harvests in the 1 L STR process had been slowly accrued over a 24 h period of perfusion, for which the vectors harvested early in the harvest cycle could have lost functionality during collection time. A possible way to resolve this lag would be to continuously process and formulate vector as it is coming off the reactor, rather than waiting for it to accumulate in a reservoir.
Another important difference between the small-scale and 1 L scale LV production processes is that in the small-scale process, cells were suspended in 50% fresh medium every 24 h after induction, whereas with perfusion the medium replacement was much more gradual, which could have contributed to the observed differences in the kinetics of vector production between the two demonstrated scales.
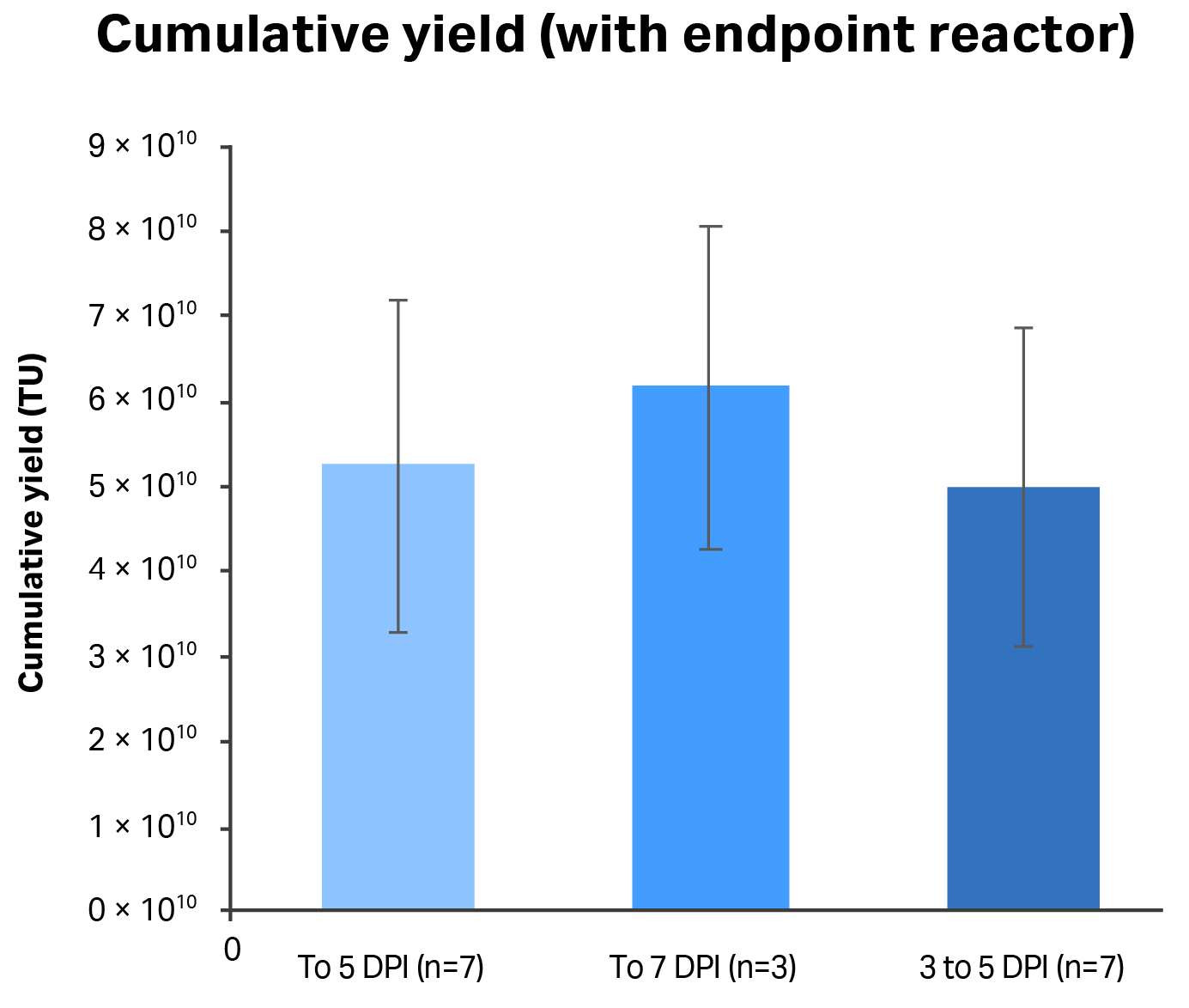
Fig 8. Comparison of different process and harvesting strategies with respect to cumulative LV yield from the 1 L STR culture process. DPI: days post-induction. n = number of runs.
Extending the 1 L STR culture process beyond five days did not increase cumulative yield of vector appreciably, nor did the inclusion of harvests from the first two days after induction (Fig 8). Increasing the culture time did not have a significant effect on yields because both cell-specific productivity and volumetric LV production was decreased by the seventh day after induction, which also resulted in reduced titers harvested from the reactor (which were about half of those observed at five days after induction). The first two days after induction yielded significantly less LV production than the next three days in all the runs, hence their inclusion in the final harvest does not significantly change the cumulative yield. Therefore, the optimal harvesting strategy was to collect three reactor volumes of harvests made at three, four, and five days post-induction and the reactor contents on the fifth day with an average cumulative yield of 5.0 × 1010 TU collected in four reactor volumes.
Conclusion
Future strategies for commercial LV manufacturing will include engineering of stable producer cells, which will simplify manufacturing, reduce costs, and ease supply chain constraints for plasmid DNA. Efficient manufacturing processes using producer cells will have to be developed and tailored to specific constraints imposed by program needs (e.g., scale), facility, equipment, and available technologies to address production bottlenecks.
In this study, we used the model LV-GFP clone 92 producer cells to develop and demonstrate a high-yielding continuous LV production process, which can be used as a basis for building processes for more clinically relevant producer cells. The process was demonstrated in 1 L STR culture with perfusion feeding and harvesting and yielded four reactor volumes of ~ 1010 TU/L of functional LV vector. This process allows continuous LV production as well as continuous harvests for up to five days. Future work may include the development of a continuous downstream processing platform to minimize the vector functionality losses associated with the intermediate storage of vector during a harvest cycle.
References
- Manceur AP, Kim H, Misic V, et al. Scalable lentiviral vector production using stable HEK293SF producer cell lines. Hum Gene Ther Methods. 2017; 28(6):330-339. doi: 10.1089/hgtb.2017.086
Regular culture maintenance
For regular maintenance clone 92 cells were cultured in HyClone HyCell TransFx-H medium (Cytiva) supplemented with HyClone Cell Boost 5 Supplement (3.5 g/L; Cytiva), 4 mM L-glutamine, and 0.1% (w/v) Kolliphor™ P 188 (Sigma-Aldrich) in shake flasks (on orbital shaker at 110 rpm, 37oC, 5% CO2 and 95% humidity) and passaged regularly when reaching densities approaching 2 × 106 cells/mL.
LVV production at 20 mL scale in shake flasks
Clone 92 cells were inoculated at a cell density of 3 × 105 cells/mL into 100 mL of HyClone HyCell TransFx-H medium supplemented with HyClone Cell Boost 5 Supplement (3.5 g/L), 4 mM L-glutamine, and 0.1% (w/v) Kolliphor P 188 and batch cultured in a 500 mL shake flask (on orbital shaker at 110 rpm, 37oC, 5% CO2 and 95% humidity). When cell density reached ≥ 5 × 106 cells/mL, cell suspension was split into 20 mL aliquots in 125 mL shake flasks. Cells were induced to produce virus by addition of concentrated doxycycline and cumate to the culture medium to achieve 1 µg/mL and 30 µg/mL final concentrations, respectively. Starting at 24 h after induction and every 24 h thereafter, cells were pelleted by centrifugation at 300 × g for 5 min, and half of the cultured medium was exchanged with fresh medium containing chemical inducers, L-glutamine, 0.1% (w/v) Kolliphor P 188, and either supplemented with HyClone Cell Boost 5 Supplement (3.5 g/L) or not. The collected medium was filtered through a 0.45 µm filter, and LV concentration was measured. Functional LV titer (TU/mL) from each of the daily harvests was assayed using the gene transfer assay on 293T cells. Daily cell counts and viability measurements were performed using the NucleoCounter™ NC-200™ after daily sampling of shake flask contents.
LV production at 1 L scale in STR
For scaling up to 1 L, clone 92 cells were inoculated at cell densities ranging from 2.5–4.0 × 105 cells/mL into 1 L of HyClone HyCell TransFx-H medium supplemented with HyClone Cell Boost 5 Supplement (3.5 g/L), 4 mM L-glutamine, and 0.1% (w/v) Kolliphor P 188 and batch cultured in the BioFlo™ 320 1 L stirred tank reactor (37oC, pH 7.15, DO 40%, and 80 rpm). When cell densities reached ≥ 5 × 106 cells/mL, cells were induced to produce the virus by addition of concentrated doxycycline and cumate to culture medium and perfused (1 reactor volume/day) with fresh medium containing inducers for five or seven days. The miniBioSep (Applikon) was used for cell retention (1 W power setting) during perfusion. Cell counts and viability measurements were performed using the NucleoCounter NC-200 after daily sampling of bioreactor contents. Functional LV titer (TU/mL) was from each of the daily 1 L harvests assayed from the seven independent runs using the gene transfer assay on 293T cells.
Gene transfer assay
The 293T cells were seeded at a density of 1 × 105 cells/well of a 24-well plate 5 h before transduction with LV. Prior to transduction, LV was serially diluted in DMEM supplemented with 8 µg/mL of polybrene and incubated at 37oC for 30 min. Transduction was performed by removing culture medium, adding diluted LV to cell,s and incubating overnight at 37oC, 5% CO2 and 95% humidity. Next day, culture medium was added to each well and cells were incubated for 72 h prior to flow cytometry for GFP expressing cells. The TU/mL was calculated using the following equation: (% of GFP positive cells [by flow cytometry] × dilution factor × number of cells seeded for transduction)/volume of transduction medium.