Extract DNA RNA and protein in one workflow for effective downstream multiomics analysis
In 1869 Friedrich Miescher performed the first DNA extraction, studying the nuclei of leukocytes. He termed the then unknown precipitate ‘nuclein’, laying the groundwork for what later became one of the key methods in molecular biology (1). Fast-forward to the current day when genomics approaches are being used often in research and clinical settings. Besides genomics, other fields such as transcriptomics have helped us to understand cellular phenotypes. And proteomics provides insights on how proteins interact and the cellular processes they are involved in. More recently, the studies and data of these individual ‘omics’ have been integrated to give rise to ‘multiomics.’ Compared with any one discipline alone, multiomics provides a wealth of information to help understand cellular processes at a much-improved resolution, giving researchers new insights into health and the mechanisms underlying disease.
Multiomics
The extraction of DNA, RNA, and protein is one of the first steps for many downstream analysis methods and diagnostic kits. These analytes can be isolated from a wide range of sample types, including fresh and frozen solid tissues, blood, saliva, cell lines, viruses, pathogens, and more. Once extracted, a myriad of techniques is available to study these analytes. Such techniques range from single sample, single mutation tests (e.g., targeted polymerase chain reaction [PCR]), up to high-throughput methods to analyze many loci or multiple complete genomes simultaneously (e.g., next-generation sequencing [NGS]).
With technological advances, we’ve seen the rise of the ‘omics’ disciplines, each generating a vast array of data. In general, the field of omics is separated into four disciplines: genomics, epigenetics, transcriptomics, and proteomics (Fig1).
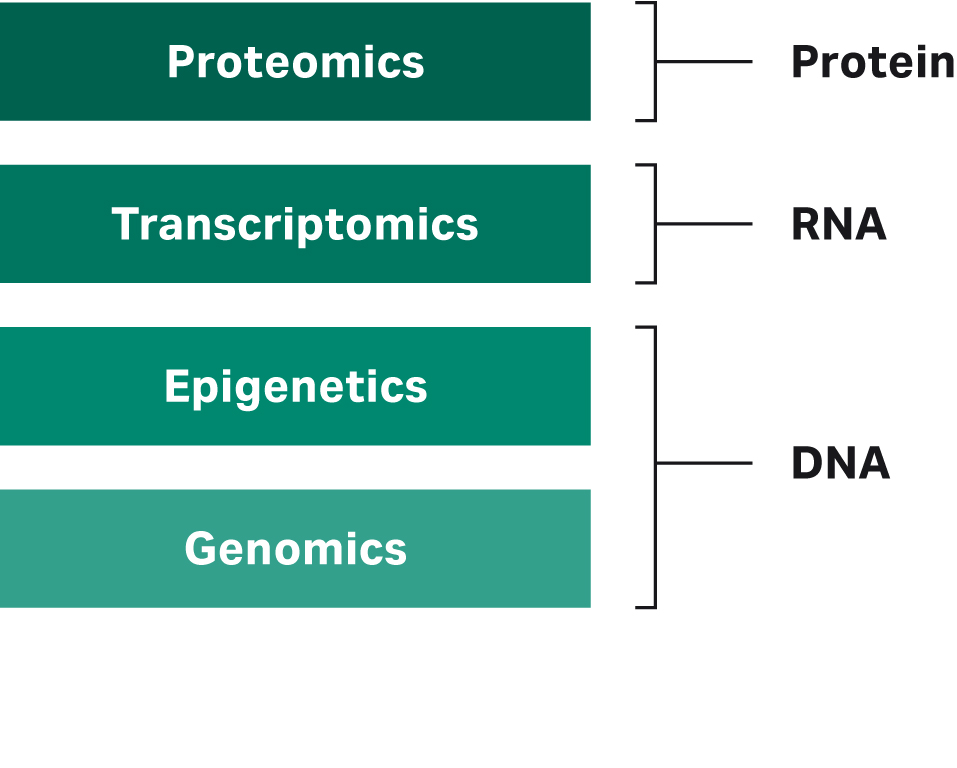
Fig 1. Illustration showing the different layers of omics and the analyte each one studies.
The DNA analyzed in genomics and epigenetics studies has identified genes, mutations, and structural information on the DNA level (i.e., in genomics) and how gene activity is controlled through methylation and histone modifications (i.e., in epigenetics). Transcriptomics studies the expression level of each gene by analyzing RNA and how the expression levels or RNA molecules themselves (e.g., due to alternative splicing) change in regulatory processes and disease. Finally, proteomics studies protein expression, which changes based on genomic and transcriptional changes and regulatory signals.
Although each of these single omics layers have given us a wealth of information, the true complexity of an organism and disease can only be unraveled by studying these disciplines in parallel, as with multiomics. According to the central dogma of molecular biology, DNA provides the genetic blueprint, which is the base code for RNA. RNA, in turn, is translated into proteins, the biological workhorses of the cell (2). To understand transcription and how the DNA is read and transcribed into RNA, both DNA and RNA must be available. Similarly, to fully uncover the pathways regulating protein translation from mRNA, both mRNA and proteins must be analyzed. A full understanding of these processes and integration of the omics layers is often needed to decipher the complex mechanisms behind diseases, which help in finding causative aberrations or treatment targets (3). These multiomics approaches have helped us further understand the regulatory processes underlying cancer, as well as infectious diseases, such as identifying the drivers for severe COVID-19 (4, 5).
Benefits of single-sample analysis
For omics analysis and any other technique that takes DNA, RNA or protein as input, analyte quality and quantity is crucial. As downstream processes become more sensitive, nucleic acid extraction can be a bottleneck, leading to insufficient or low-quality data downstream. A survey by the National Cancer Institute among cancer researchers found that the lack of quality biospecimens resulted in 81% of researchers having to limit the scope of their research (6). This problem is exacerbated in multiomics studies, which not only require high quality for all analytes, but often also require more material, as each analyte is extracted from a separate sample.
One way to overcome these issues is by extracting all the analytes from the same sample. Although most extraction kits focus on extracting either DNA, RNA, or protein, the TriplePrep extraction kit is designed to extract all three analytes with high yield and quality from the same tissue in a single workflow (Fig 2). The TriplePrep workflow starts with homogenization of the sample, followed by a DNA purification process. The flowthrough from DNA purification contains RNA and proteins, which can be captured in subsequent steps; RNA first, and then the flowthrough can be used to precipitate and resuspend proteins. The final result will be up to three tubes, depending on which analytes are needed. Each tube will contain a high-quality, high-yield elution of the purified analyte from the same cell line or tissue sample (Table 1).
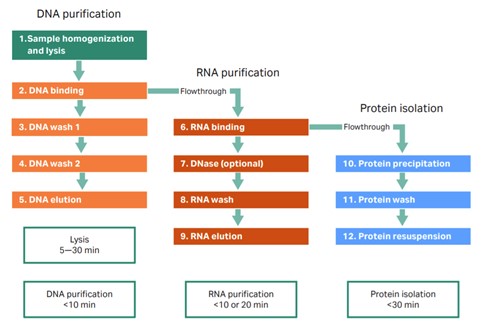
Fig 2. TriplePrep workflow. From sample to DNA,RNA, and protein in less than 60 min.
Table 1. Typical yield and purities of genomic DNA, total RNA, and total protein from various tissues
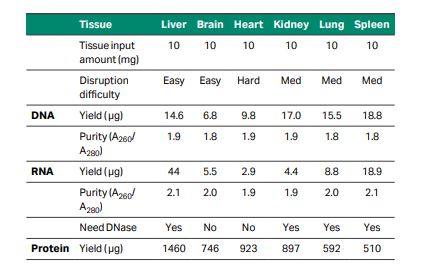
Reducing the need to use multiple samples to extract DNA, RNA, and protein is especially helpful when working with limited samples from, for example, biobanks or small biopsies. Another benefit of extracting analytes from the same sample is improved accuracy of results. With methods that extract one analyte, a sample from a tumor or tissue is split into two or more pieces; each is processed to yield the analyte for downstream processing. One complication with splitting a tumor or tissue is that it must be homogenous to allow for accurate deductions when combining the DNA, RNA, and protein data downstream. Even within a single cultured cell line, substantial heterogeneity exists (7). Tissues, especially tumor tissues, are often highly heterogeneous. This heterogeneity makes it challenging to link the findings from the DNA from one piece of tumor to the RNA from another part and use this data to infer a link between the two (8). With TriplePrep, all analytes are derived from the same sample, which minimizes these challenges and allows for more meaningful data and analysis linking the findings from the different analytes (Fig 3).
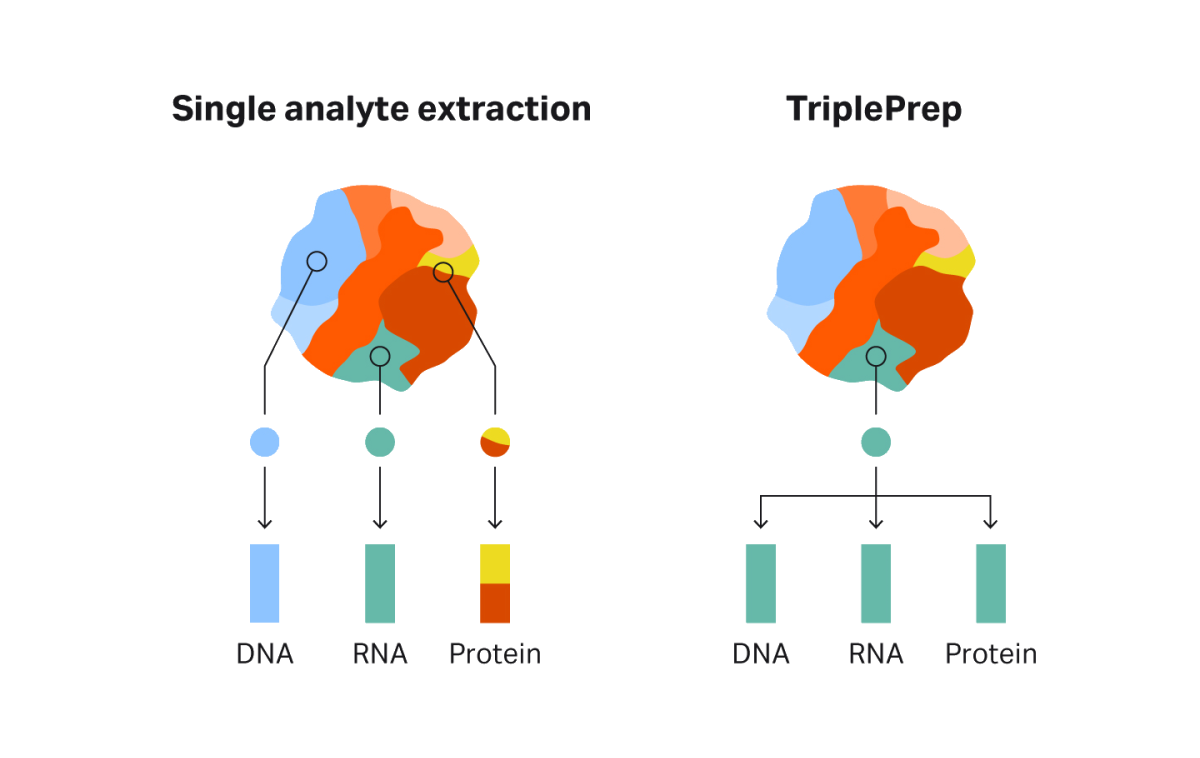
Fig 3. Tissues can be highly heterogeneous, making it challenging to link analyses from DNA, RNA, and protein derived from different parts of the tissue. With TriplePrep, all data comes from the same tissue sample, allowing meaningful deduction of regulatory processes between the analytes.
Genomic DNA, total RNA, and total denatured proteins isolated with TriplePrep can be used for common genomic and proteomic techniques. Examples include PCR, restriction enzyme digestion, next-generation sequencing (NGS), array comparative genomic hybridization (CGH), real-time polymerase chain reaction (RT-PCR), Western blotting, 2-D difference gel electrophoresis (DIGE) and more. Thus, researchers can choose from a wide range of analysis methods to interrogate their sample of interest (Fig 4).
A
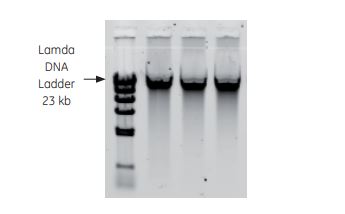
B
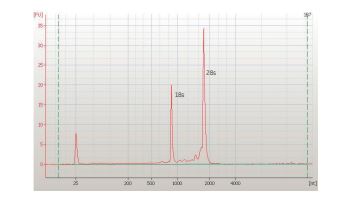
C

Fig 4. Examples of downstream analyses with TriplePrep. A) Genomic DNA from 1 million HeLa cells evaluated using 0.8% agarose get electrophoresis. B) Total RNA from 10 mg rat liver evaluated using the AgilentTM 2100 bioanalyzer showing high-quality RNA (RIN: 9.8). C) Total protein from 10 mg rat liver evaluated using 2-D DIGE. Overlaid image of TriplePrep (yellow) and reference gel (red), showing more spots are detected with TriplePrep (green spots).
Summary
Multiomics enables scientists to investigate samples at improved resolution compared with single omics approaches. Thus, multiomics aids our understanding of health, disease, and development of therapies. At the heart of the methods used in these studies lies analyte extraction and the ability to link the results together in downstream analysis. With TriplePrep, researchers can extract high-quality, high-yield DNA, RNA. and protein from the same sample in a streamlined workflow. The kit provides excellent starting material for their experiments, resulting in accurate and meaningful results.
References
- Dahm R. Friedrich Miescher and the Discovery of DNA. Amsterdam, The Netherlands: Elsevier; 2004.
- Crick F. Central dogma of molecular biology. Nature. (1970);227:561-563. doi: 10.1038/227561a0.
- Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18(1):83. doi:10.1186/s13059-017-1215-1.
- Lu M and Zhan X. The crucial role of multiomic approach in cancer research and clinically relevant outcomes. EPMA J. 2018;9(1):77-102. doi:10.1007/s13167-018-0128-8.
- Carapito R, Li R, Helms J, et al. Identification of driver genes for critical forms of COVID-19 in a deeply phenotyped young patient cohort. Sci. Transl. Med. 2021;14(628):abj7521. doi: 10.1126/scitranslmed.abj7521.
- Massett HA, Atkinson NL, Weber D, et al. Assessing the need for a standardized cancer HUman Biobank (caHUB): findings from a national survey with cancer researchers, J. Natl. Cancer Inst. Monogr. 2011;(42):8-15. https://doi.org/10.1093/jncimonographs/lgr007.
- Tang L, Yang M, Mueller T, et al. Investigating heterogeneity in HeLa cells. Nat. Biotechnol. 2019;(16):281. https://doi.org/10.1038/s41592-019-0375-1.
- Litchfield K, Stanislaw S, Spain L, et al. (2020) Representative sequencing: unbiased sampling of solid tumor tissue, Cell Rep. 2020;31(5):107550. doi: 10.1016/j.celrep.2020.107550.