
Frost & Sullivan recently invited industry leaders with viral vector experience in cell and gene therapy to participate in a new thought leadership forum, a Virtual Think Tank (VTT) series. This forum brought together leading minds in this emerging field to discuss the current state of regulatory issues around viral vector manufacturing, key challenges, and other insights related to viral vectors. The key opinion leaders (KOLs) who contributed to the discussion included:
- Sharon Turner-Rinehardt, Director of Regulatory Affairs, Catalent, Paragon Gene Therapy, Inc.
- Mats Lundgren, PhD, Customer Applications Director, Cytiva
- Bob Kozak, Senior Director, Regulatory Affairs, Bayer Healthcare Pharmaceuticals
- Karen Magers, Head of Regulatory Affairs Cell and Gene Therapy Technologies, Lonza
Gene therapies transfer genetic material to a patient to modify a gene triggering a disease, with the goal of effectively treating that disease. Gene therapy development and production is a complicated endeavor that involves collaborative input from academic, industry, and government entities. Numerous global regulatory agencies have published guidelines and regulations directing this type of work (Fig 1), but not all guidelines are aligned.
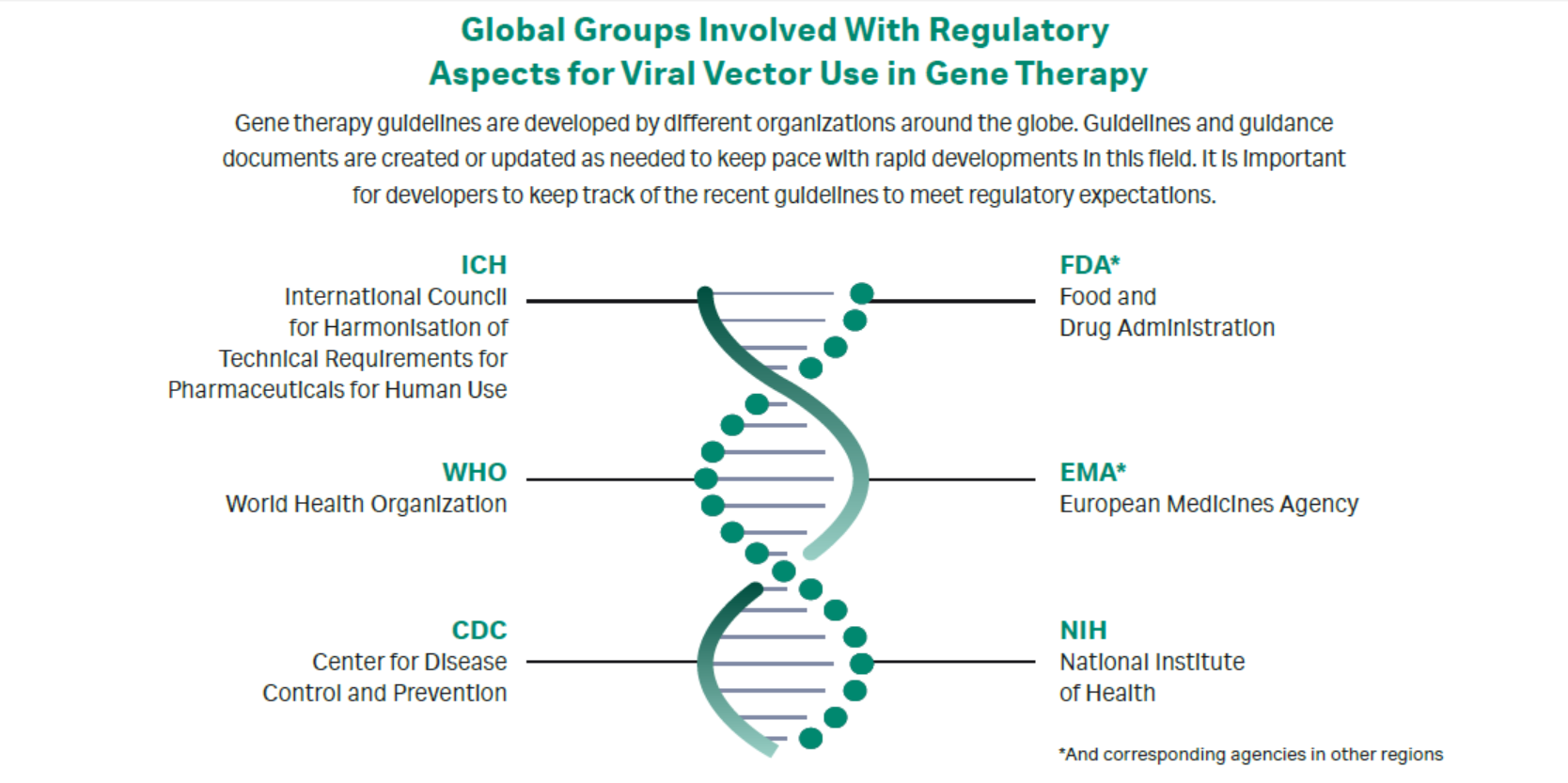
Fig 1. Global groups involved with regulatory aspects for viral vector use in gene therapy.
Advances in gene therapy and the introduction of genetic material using viral vectors come with a therapeutic benefit. However, there are challenges on the development side. Viral vectors differ significantly with regards to their virulence and replication status. Viral vectors can be:
- integrating (e.g., retroviral and lentiviral vectors), which incorporate a specific transgene into the host’s chromosomal DNA, or
- ·non-integrating (e.g., adenoviral, herpes, and AAV vectors), which do not assimilate the desired transgene into the chromosomal DNA but instead leave the transgene inside the nucleus (1).
Viral vector production is complex. For one thing, legacy technologies adopted from the academic research side are hard to scale up. Also, technology challenges coupled with talent shortages are a further bottleneck to expanding capacity. The relatively young cell and gene therapy market is highly fragmented globally. Few contract research and manufacturing organizations (CMO/CROs) are commercially scaled to assist with viral vector demands and shortages, and only a handful of the larger companies are capable of commercial-scale production. Finally, solutions that allow companies to expand their own capacity are limited.
Guidances from the National Institutes of Health (NIH) and Centers for Disease Control (CDC) are not well defined. The Food & Drug Administration (FDA) and the European Medicines Agency (EMA) guidelines for Good Manufacturing Practice (GMP) regulations related to regenerative medicine development workstreams are helpful, but they differ between agencies and are not fully compatible. These differences can confound companies making and marketing products globally (1).
Challenges facing the field of gene therapy are not trivial. Of note, manufacturing of viral vectors is far from straightforward. Companies have reported capacity issues, incongruent regulatory guidelines between the FDA and EMA, as well as insufficient numbers of trained workforce available. Taken together, these challenges interfere with the ability to produce the quantities needed for clinical trials. In response, some companies have built in-house manufacturing, which gives them more control over their supply and strategies. Most companies aim to use technologies that enable cost-effective and scalable production, which requires the use of efficacious clinical-grade viral vectors.
The FDA permits incremental collection and reporting of biologic information. For instance, assay potency is reported at Phase 1, and validated, qualified assays are reported at Phase 3. The EMA, on the other hand, requires full GMP compliance prior to the start of the development of clinical programs. Further, when situations arise where full compliance with their guideline is not possible, the EMA requires proper justification and explanation of the alternative approach for obtaining comparable information (3). These differences in reporting requirements also apply to viral vector-specific guidelines.
The current trend for biotech and pharma companies is to require full GMP compliance of their academic partners, even in the early phases of clinical development. The Investigational New Drug (IND) application process drives this trend. From a business perspective, there is a need to lower the risk as the products move through development toward commercialization.
The FDA is optimistic about the potential for gene therapy-driven treatments. In an effort to simplify some of the regulatory oversight, the agency has issued several guidances, including two in early 2020.
Expectations for regulatory processes
Regulatory compliance is top of mind for those who use viral vectors. There is uncertainty around the regulatory challenges, as both regulators and companies are often new to the emerging gene therapy area. Kozak noted, “There is a lot of support by the FDA as far as guidance and recommendations that people can rely on, and the NIH guidelines for safety and containment also provide a lot of background.”
Some of the more noteworthy FDA guidances mentioned during the discussion included:
- 1998 guidance addressing vector construction, characterization, and production systems
- 2012 preclinical guidance
- 2016 guidance on microbial vectors
- 2020 guidance, including document on testing of replicative competent vector virus testing
- 2020 guidance on Chemistry, Manufacturing and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs)
Many of the guidances apply to viral vectors.
Magers and Kozak remarked that the FDA guidance on CMC information for INDs is highly prescriptive and includes detailed advice. Several panelists mentioned the need for more phase-appropriate recommendations for the IND and manufacturing.
The EMA guidelines that have been issued for public consultation, including the guideline on quality, non-clinical and clinical requirements for investigational advanced therapy medicinal products in clinical trials “are not as detailed as the FDA’s guidances,” said Magers.
The EMA has used its PRIME designation to accelerate the development of gene therapies. The PRIME designation supports the development of medicines targeting unmet needs, offering early and ongoing interaction with the agency to smooth the path forward.
Both Kozak and Lundgren reported that there is much activity outside the US and Europe for implementing accelerated programs for cell and gene therapy. For example, Japan is active in the gene therapy area and is working hard on developing regulations. The nation has developed innovative manufacturing technologies. Also, it has a cell-based therapeutics products group in its Ministry of Health, MHLW, which has oversight on gene therapy.
Several panelists noted that on an international level, the International Conference on Harmonization (ICH) and World Health Organization (WHO) are interested in developing harmonized guidances that may be interpreted internationally. There is concern within the industry that the regulatory predictability and harmonization might not be achieved. At a high level, the FDA, EMA, ICH, and WHO are not classifying gene therapies in the same manner, which will make cross-applicability of the guidance documents difficult. The terminology differs, as does the definition of gene therapy.
“Harmonization of guidances will be key,” said Turner-Rinehardt. She noted “the importance of the worldwide regulatory agencies coordinating their efforts. Companies are concerned about being able to manufacture in the US and also about being able to produce and distribute their products in other countries. Harmonizing guidances is going to be of key importance going forward.”
Challenges for viral vector manufacturing
The transition from academic laboratory to a GMP capable facility can be difficult for some products. The panel agreed that it is not a simple task to go from development assays to validated, releasable assays that are easy to use and affordable. Some of the larger groups have the assays they need, but for those that don’t, it is difficult to catch up. With CMO/CRO capabilities maxing out, it’s not easy to get help developing the analytical assays. Companies that remain focused on things like potency do not have time to make all the other validated assays necessary for release. Finally, the panel noted that there is a struggle to find a sufficient number of trained workers to help get all the assays ready for launch.
Dr. Peter Marks, Director of the Center for Biologics Evaluation and Research (CBER) (4), has been vocal about the challenges faced with gene therapy development. He recognizes that with over 1000 gene therapy clinical trials underway globally, it is difficult to have enough viral vector production capacity to keep up. Lundgren concurred and noted that companies sometimes resort to developing their own manufacturing sites to generate their own vectors.
There is a plethora of programs coming from academia, where inventions of new products happen regularly without the capability for reproducible manufacturing at large scale. Both Magers and Turner-Rinehardt noted that the FDA is concerned about controlling for contamination and cross-contamination, because each vector will not necessarily have its own fully segregated and contained facility.
Lundgren added, “If there is closed processing in terms of the equipment and disposables, then at least you are going in the right direction.”
Kozak said, “It is still very difficult but may be the way to go; if you have closed systems, with sampling strategy, separate air handling systems are important elements to consider. There is an advanced technology team (ATT) in place, CBER (CATT) and, separately, an emerging technology team (ETT) in place at CBER (CETT) to discuss facility designs and how manufacturing can be done and to address the concerns of cross-contamination and safety of products.”
Lundgren further elaborated, “There are other challenges around the analytics and how you are going to analyze all these vectors. How do you put control into manufacturing based on analytics? There are quality-by-design (QbD) concepts, but they are not so easy to implement when it comes to viral vectors. Whether the empty capsids are immunogenic or not and to what extent you need to have pure vectors to move forward for in vivo gene therapies remains a bit of a controversial topic.”
One of the biggest challenges, noted Magers “is the CMC development buried below the footprint of the Phase 1, Phase 2, and Phase 3 trials. The diseases these drugs are in development for are so rare, such ultra-orphan indications, that the total registration trials is 10 to 20 participants. It is a challenge to be able to do all the things that are required for commercialization in the CMC space with such an accelerated clinical trial program.”
There is an urgent need to resolve the technical and capacity issues to get these therapies to patients as quickly as possible. The panel agrees that CMC is a rate-limiting step.
Regulatory scope of gene therapy
The regulatory scope for gene therapy for various vector applications, such as using a lentivirus as an agent to transduce the patient cells ex vivo versus in vivo, is an issue that goes back to classification. A cell-based product that is genetically modified ex vivo can be called gene therapy.
Magers elaborated, saying, “In the EU, the viral vector would be considered a starting material. That is a major disconnect with how the US is regulating the material that is used to make the drug. In the US, we are calling the starting materials used to manufacture the product one thing, but the viral vectors something else. That point came out of the 2020 guidance directing that the information required for a viral vector be analogous to the drug substance.”
“The expectations for safety evaluations require three pillars of standard tests which are done on traditional biologics,” said Kozak. “In contrast, with gene therapies you have to be focused on the careful selection of raw materials and testing them to be free of adventitious agents. Some of the purification methods can lend themselves to clearance of potential adventitious agents where others cannot. Further, it is very important for AAV development that the companies evaluate their purification process for viral clearance opportunities. The chromatography steps may have various clearance capabilities. For AAV we are lucky that it’s non-enveloped and robust. It is harder for lentiviruses that are enveloped and very sensitive. We don’t know what other methods are available to get rid of potential pathogens. It is challenging and requires more regulatory authority and review.”
Lundgren agreed that lentiviruses are highly unstable, adding, “It is difficult to have some kind of virus clearance in the purification process. You have to rely on cell line testing, raw material safety, and all the containment and closed processing. It is very similar to production of live viral vaccines in that respect.”
Regulatory scope regarding viral vectors and plasmids
Plasmid DNA is a raw material in gene therapy production. Kozak remarked that “plasmid production is typically done through recombinant E. coli fermentation. In the fermentation process, genetic sequences are amplified, harvested, purified and then tested for safety. Significant challenges exist with lot-to-lot consistency and variability and purity of the resulting product. Regulations dictate that the end product needs to be greater than 95% pure plasmid DNA that is free of process-related variants and impurities. For viral vector production, cell banks are created. Fortunately, cell banks can be screened and characterized as either the producer or as the plasmid generator. All of this can be controlled with safety guidelines in place. That would be part of the normal description of what is anticipated as part of the package.”
Magers said, “The requirements for plasmid DNA are also not completely clear. It is more black and white if the plasmid DNA was actually the product, but plasmid DNA can also be a starting material for generating the viral vector. The phrasing and terminology in the guidance documents states that these types of starting materials should follow GMP principles.” The panel agreed that if you start with a plasmid that is not under GMP conditions, it is tough to understand the safety aspects of not producing in that environment. A lot of questions were raised: What are the long-term effects? Is there a test or are controls in place to be able to monitor that? The panel concurred that the FDA or the regulatory agencies in other countries should have a more standardized recommendation or guideline around these aspects.
Another point of agreement was the gray area where people can decide whether to produce plasmids used as starting materials in a GMP environment. If it is a critical raw material, it must have a high quality. When plasmids are used for generation of viral vectors and the drug itself, GMP is necessary. Regulations need to offer clear guidance.
According to Kozak, seed stocks used for the plasmids must be tightly controlled, so their sourcing and derivation is described. These stocks are used to generate the final producer cell lines in the characterization of the cell bank. Any safety issues should be covered and safety testing done before the cells are used.
The cell lines can be tested once they are generated, Lundgren pointed out, adding that the problem here is probably for the companies using huge amounts of plasmid DNA for transient transfection when producing viral vectors. Magers stated the need to stay as close to GMP as possible. A large volume of vector will be generated, and the controls around GMP are based on ensuring safety and consistency as well as having the correct documentation.
Kozak added, “CBER offers a lot of pre-IND meetings where these kinds of things can be discussed. You can identify what extent you have to have cell banks tested and expectations of clonality. You can bring things up early on with the health authorities to make sure you are both on the same page. If there are safety concerns, they can be addressed, avoiding FDA putting the IND on hold because of the issue.”
Regulatory requirements and harmonization
Regulatory requirements and harmonization are topics that a lot of regulatory bodies are dealing with right now. There is excitement about the products being developed at accelerated rates, with some products having the potential to be clinical cures. The panel concurred that international health authorities must be able to assess these programs so that, at the global level, patients will have access to these existing products. Both the WHO and ICH are just getting started with the harmonization. It is not a fast process but one that must ensure that regional changes can be adopted globally (Fig 2). It is frustrating when there is no mutual recognition and reliance programs are too complicated or not used.
Fig 2. Regulatory collaboration is key to improve global access.
Global harmonization is a big issue. Health authorities may not have the capacity or the resources to review some of the new modalities. US regulators must figure out what kind of solutions will work, so they can be shared with the international authorities. If one international reference body strategically cooperates with US regulators and signs off, other regions might accept the decisions of a reference body to accept and approve products, which would be ideal. Then, within six months to a year after something was approved in the US or Europe, it could possibly be rolled out globally. Kozak noted, “With a limited workforce and the level of expertise required, this may take time to develop a system analogous to the EU centralized procedure model for cell and gene therapy products.”
Conclusion
Viral vector manufacturing is a critical part of developing and producing gene therapies that use them. Since manufacturing custom-made viruses can consume over a third of a biotech company’s R&D budget, decisions that improve manufacturing efficiencies, such as using the services of a CDMO, will have a potential impact on patient access and, ultimately, drug costs.
Despite the variation in regional guidelines and talent shortage, gene therapy research is moving forward. But without harmonized guidelines, global efforts will continue to be hindered and will likely result in redundancy of efforts, including in processes where viral vectors are used. At the end of the day, companies will have to decide whether to build their own clinical or commercial-scale manufacturing capabilities or continue to rely on CDMO/CRO partners with the resources and capacity to meet their current clinical needs.
Frost & Sullivan would like to thank the cell and gene therapy thought leaders that joined the Virtual Think Thank for their time and valuable insights into this promising field. We hope that this discussion spurs new ideas and fosters additional exchanges for this burgeoning field.
Tips for getting your investigational drug regulatory ready.
Learn about Cytiva’s resources and support for gene therapy development and manufacturing.
References
- Ehrhardt A, Xu H, Kay MA. Episomal persistence of recombinant adenoviral vector genomes during the cell cycle in vivo. J Virol. 2003;77(13):7689-7695. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC164819/
- Petrich J, Marchese D, Jenkins C, Storey M, Blind, J. Gene replacement therapy: a primer for the health-system pharmacist. J Pharm Pract. 2019;6:1-10. https://pubmed.ncbi.nlm.nih.gov/31248331/
- European Medicines Agency—Committee for Advanced Therapies (CAT). Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products. 22 March 2018; EMA/CAT/80183/2014. https://www.ema.europa.eu/en/quality-preclinical-clinical-aspects-gene-therapy-medicinal-products. Accessed May 26, 2020.
- FDA Organization, Dr. Peter Marks, FDA’s Efforts to Advance the Development of Gene Therapy. (2019) May 1. https://www.fda.gov/news-events/fda-voices-perspectives-fda-leadership-and-experts/fdas-efforts-advance-development-gene-therapy. Accessed May 26, 2020.