Article inspired by a Tapas and TECH Talks digital event with insights from customer collaborations. Experts were from Cytiva and CCRM.
This article discusses options for cell therapy process development and validation that minimize risks through quality control (QC), optimization, scaling, closing, automation, and assay development, all in order to prepare for successful commercial production.
Cell therapy scale-up strategies
To achieve commercial success, a developed process must be scalable and suitable for a manufacturing environment. The scaling strategy depends on the type of cell therapy. Allogeneic therapies are meant to serve many patients. This scaling strategy of traditional bioprocessing, such as monoclonal antibody therapy processes, commonly uses a seed-train approach, where you scale across systems, progressively increasing in size. For allogeneic cell therapies, in order to minimize sterility and handling risks in more precious patient-derived cells, scaling within the same system is desirable. This can be done with low cell volumes, starting with batch feeding, and then ramping up feed and waste production rates with perfusion to progressively expand cells. Also, in contrast, autologous cell therapies are personalized for each patient, so the batch sizes are small. Autologous therapies use a scale-out strategy, where additional small bioreactors are added to increase production throughput.
Allogeneic processes can be optimized at scales from 100 mL to 2 L, and volumes up to 25 L can be achieved in Cytiva’s Xuri rocking bioreactors. Very large batch needs up to 2000 L can be met in Cytiva’s Xcellerex line of stirred-tank bioreactors. For autologous processes, the 0.5 to 5 L volume range is usually enough. Large-scale bioreactors can be used for other immunotherapy platforms that require feeder layers or stimulatory cell lines.
Modern scale-up strategies for CAR T cell manufacturing, specifically the expansion of suspension T cell cultures, are very amenable to transfer into bioreactors for expansion. This allows the use of single-use equipment with closed connections, preferably sterile, weldable tubing lines, as well as automated liquid handling and perfusion. These advantages greatly lower the risk and cost compared with manual, labor-intensive processing while minimizing operator manipulations.
CAR T cell process development
To validate a process and achieve consistent production of a biologic, you must maintain your ranges of critical process parameters (CPPs) and critical quality attribute (CQAs) throughout the process. In order to do this the CQAs and CPPs must first carefully be defined and prioritized (1).
In addition to process development, minimizing variation in your process is key. For cell therapies, being able to measure variation within a process, consistently across patients, requires the right analytics.
Tip: In early development, heavy characterization with many measurements can help you to pinpoint the sources for variation and really define your CQAs and CPPs and their ranges. Assay acceptance criteria can also be built around these to control for consistent desired CQA/CPP targeted outputs.
To develop final process controls and lot release assays, you must follow regulatory requirements, of course, and incorporate any specific logistical needs to your process. The ideal state is to be able to demonstrate control and to have the analytics that use nondestructive sampling by incorporating tools and technologies with in-line, real-time monitoring, wherever feasible. Ultimately, if you have a flexible process in which real-time monitoring allows parameters to be adjusted, you can simplify process control and ensure each patient batch is able to hit the target therapeutic dose and characteristics.
Process development decisions
Table 1 summarizes the key options to consider when developing commercial-scale manufacturing of a CAR T cell process.
Table 1. Main decisions for CAR T cell manufacturing at commercial scale
| PD focus area |
Best practices |
Benefits |
||||
| ↑ Product development |
↑ MFG operations |
↓ Process variability |
↓ Production risk |
↓ Regulatory burden |
||
|
Characterization |
Small-scale optimization | x | x | |||
| Develop meaningful CQAs and CPPs | x | x | x | x | ||
| Characterize high-risk source for process variability | x | x | x | x | ||
| Correlate CQAs with variation to define process tolerance | x | x | x | x | ||
|
Raw material selection |
GMP reagents and media formulation screening | x | x | x | ||
| Define ancillary reagents | x | x | x | |||
| Obsolete poorly defined raw materials | x | x | x | x | ||
| Identify supplier sourcing with backups | x | x | ||||
|
Cell or gene input material |
Define input cell source selection and availability | x | x | x | ||
| Optimize T cell enrichment and isolation strategies | ||||||
| Viral vector selection and titer optimization | x | x | x | x | ||
|
Final process |
SOP generation with built-in unit op tolerances | x | x | x | x | |
| Engineering runs with integrated unit ops | x | x | ||||
| Tech transfer to CMO for process validation | x | x | ||||
| Process validation activities | x | x | ||||
Each process development aspect has different benefits that may drive your focus areas. Larger investments in process development can increase your chances for commercial production successes, and many of these benefits also imply a reduction in cost of goods (COGs). But ultimately, this must be balanced with the race to get through clinical trials. We highly recommend closing and automating your manufacturing process. Different processes have different needs, and solutions need to be tailored to suit and balance the required costs and timeline. Cytiva's Fast Trak process development services can help you to implement best practices in areas where you want to focus.
Figure 1 represents a cost savings prediction for mesenchymal stem cells (MSCs), if you were to fully close and fully automate the process (2).
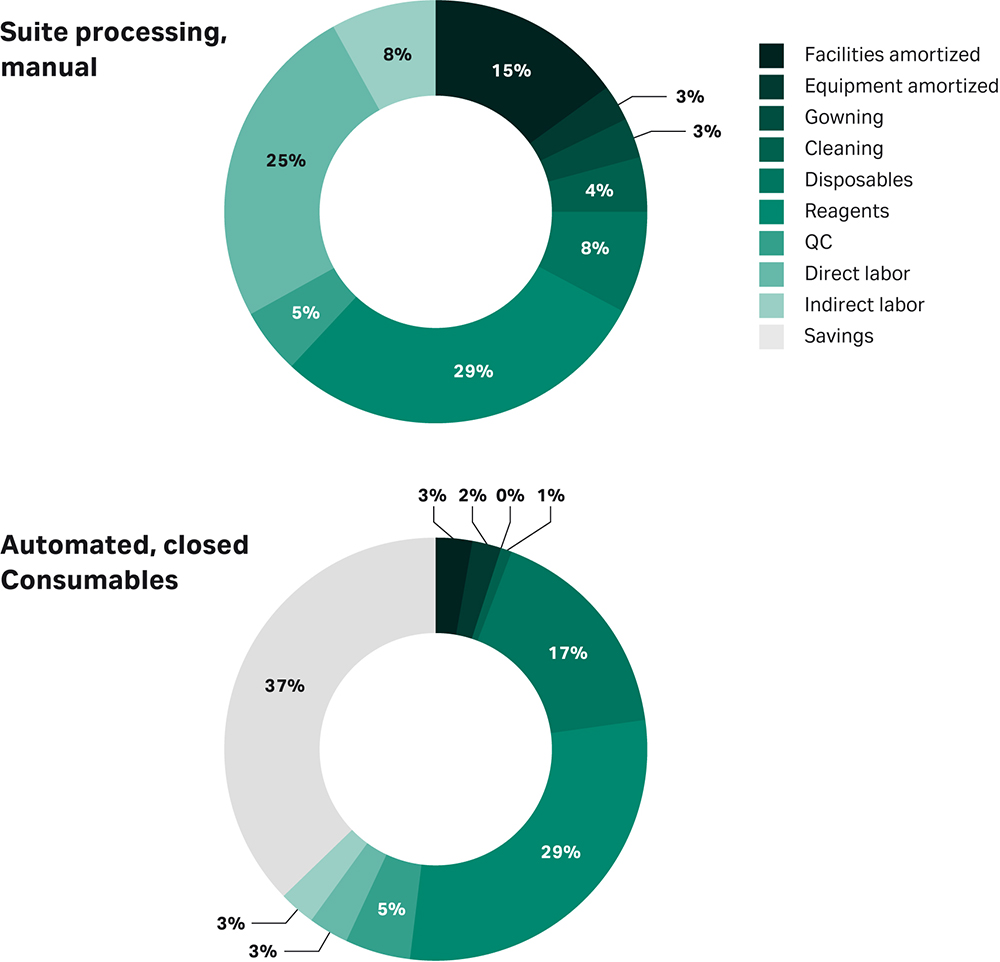
Fig 1. Predicted costs of a manual vs closed and automated cell therapy manufacturing process. This example is for MSCs based on Lipsitz et al. Cytotherapy, 2017.
The earlier you invest in closed and automated processing, the easier the process optimization will be, and the quicker tech transfer to start clinical trials will be. And, of course, you simplify commercial operations. Automation allows parameters to be adjusted through controlled and traceable digital inputs in the equipment, in contrast to error-prone manual processing. Also, fully closed processes clearly reduce contamination risks while allowing you more flexibility in logistics in your facility requirements.
Quality control and release testing
How do the regulators feel about the importance of monitoring how our manufacturing processes perform? They use specific language and terminology around the identity, strength, quality, purity, and potency. With this in mind, as well as the fact that the clinical trials start with an initial focus on safety, we can define the elements required.
Other important aspects from the regulatory point of view are method qualification and method validation. The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) has a good, rich set of guidelines. Complementing those are the United States Pharmacopeia (USP) chapters on validation, verification, and method transfer. If you are marketing in the US, you will definitely need to review those as well as specific Food and Drug Administration (FDA) guidances. Table 2 lists the key guidelines, and Table 3 summarizes the regulatory categories and activities required for Phase 1 and 2.
Table 2. Key regulatory guidelines for cell therapies marketed in the US
| Agency |
Guideline |
| ICH | Q2(R1) Validation of Analytical Procedures |
| USP | <1225>: Validation of Compendial Methods |
| USP | <1226>: Verification of Compendial Methods |
| USP | <1224>: Transfer of Analytical Procedures |
| FDA | Analytical Procedures and Method Validation for Drugs and Biologics (2015) |
| FDA | Bioanalytical Method Validation Guidance for Industry (2018) |
Table 3. Summary of regulatory categories and activities required for Phase 1 and 2
| Regulatory category |
Source of testing material |
Example |
Activity required (Phase I/II) |
| Safety/Purity | Compendial | Sterility, USP <71> | Verification |
| Endotoxon, USP <85> | Verification | ||
| Mycoplasma, USP <63> | Verification | ||
| Impurity testing | Qualification/Validation | ||
| Identity | Process development | Flow cytometry | Qualification/Validation |
| Karyotype | Qualification/For information only | ||
| STR | Qualification/For information only | ||
| PCR methods | Qualification/Validation | ||
| Strength | Process development | Cell counting/viability | Qualification/Validation |
| Quality | Process development | Performance test | Qualification/Validation |
| Proliferation testing | Qualification/Validation | ||
| Animal models | Qualification/Validation | ||
| Potency | Process development | Flow cytometry | Qualification/Validation |
For safety and purity, these methods are compendial and apply across the board to almost all products. So, only verification will be required for sterility, endotoxin, and mycoplasma. However, any process-specific impurities will need qualification and validation.
Process robustness
Process robustness is an important manufacturability parameter, to make sure that the process will work not only in a perfect research and development space but also during the normal repeatable conditions of manufacturing operations. For example, if samples have to be collected and tested immediately, does that mean lab analysts have to work 24 hour shifts, when that sample is pulled? The goal is to perform a robustness study and demonstrate stability of the sample on the bench, in a fridge, or in a freezer, until morning when it can be analyzed on a scheduled timetable.
Tip: Qualification is actually a subset of method validation, before Phase III. Eventually, these methods will need a full validation, so if early indications show that there may be issues that would impact a validation, a new strategy may be required.
Summary
We have described the requirements to get a process ready for manufacturing. And while manufacturing process validation is not required for an early phase project, it does apply for the analytics in the early phases.
Tip: It is so important to make decisions when designing a process, so that there is a pathway to guide your decision making throughout the development life cycle.
Process qualification is always designed and evaluated to determine and reduce variability as much as possible, in order to support the end goal of commercial manufacturing. And when you’ve completed all this development work, you still monitor and continue to improve the variability and reduce failure risk during the product life cycle and as projects go forward.
Most cell therapies still use risky and expensive open, manual processes. The goal is to move towards closed and automated equipment in order to reduce costs and risk, as well as to modernize to meet regulatory and supply demands.
References
- Lipsitz YY, Timmins NE, Zandstra PW. Quality cell therapy manufacturing by design. Nat Biotechnol. 2016l34:393-400. doi: 10.1038/nbt.3525.
- Lipsitz YY, Milligan WD, Fitzpatrick I, et al. A roadmap for cost-of-goods planning to guide economic production of cell therapy products. Cytotherapy. 2017;19:1383-1391. doi: 10.1016/j.jcyt.2017.06.009.