Key takeaways:
- Early decisions determine scalability and GMP readiness. Choices made during research can create significant barriers when processes must be standardized, documented, and scaled.
- Consistency is essential for clinical translation. As programs move toward clinical manufacturing, reducing variability becomes as critical as maximizing yield for regulatory compliance and scaling.
- Processes outside the manufacturing facility are as critical as those within it. Conditions for apheresis, cryopreservation, transport, and thawing directly impact dose viability and consistency.
What you will learn
This guide explains the end-to-end manufacturing process for autologous CAR T therapies, with a focus on the process decisions that matter when moving beyond the bench. It highlights where variability, scalability, and compliance challenges emerge, and how teams can plan for them early.
Autologous chimeric antigen receptor (CAR) T cell therapies have earned their spot in the history books as a groundbreaking approach to cancer treatment. While the idea of engineering a patient’s immune cells to target and destroy their own malignant cells may sound like science fiction, CAR T therapies have made a very real impact on the lives of thousands of patients, and even more family members.
Behind each dose lies a complex and tightly controlled manufacturing process. It begins with isolating immune cells from a patient’s blood and ends with the formulation of a personalized therapeutic product. Every step must be executed with precision to ensure safety, efficacy, and consistency.
This guide walks through the end-to-end workflow of autologous CAR T manufacturing, highlighting key steps such as T cell isolation, genetic modification, expansion, and final formulation.
Along the way, we’ll uncover the technical challenges inherent to each step and explore how the industry is addressing them through innovative equipment, automation, and process design strategies to help ensure every patient has access to the therapy they need, when they need it the most.
Collecting patient samples for CAR T cell manufacturing
As an autologous treatment, CAR T therapy production starts at the patient. Apheresis is the targeted separation of blood components to collect certain fractions, while leukapheresis is specifically optimized for harvesting white blood cells, including T cells, for cell therapy production. The collected cells are packaged, often cryopreserved, and then transported to a manufacturing facility for further processing.
Early-stage teams often treat this step as a clinical formality, but in reality, the quality of the apheresis product sets the ceiling for achievable yield, cost, and timelines. Even a well-designed manufacturing workflow cannot fully compensate for poor starting material.
Patient factors such as disease state, immune suppression, and prior treatments can influence the quality of the apheresis product in terms of cell count, cell viability, composition, and more.
Managing starting material variability from the clinic to manufacturing
Although patient biology cannot be changed, there are controllable factors that manufacturers should keep in mind to minimize variability of the starting material. Standardizing apheresis protocols, implementing clear transport and handling requirements, and adopting consistent documentation practices all help prevent quality loss between the clinic and manufacturing site.
Go deeper:
Separation and isolation in CAR T production
Once the patient cells arrive at the manufacturing site, the first task is to isolate T cells from other blood components to obtain a highly pure, viable population for downstream gene modification and cell expansion.
T cell isolation is one of the first opportunities manufacturers have to combat starting material variability and maximize the potential of later expansion steps. Teams frequently underestimate how variable leukapheresis is, and how this variability directly influences efficiency of downstream processes—and ultimately the ability to deliver a viable therapeutic dose.
Common sources of variability include:
| Source of variability | What changes | Why it matters downstream |
|---|---|---|
| Patient baseline and disease state | Absolute CD3+ counts; proportion of naïve/central memory T cells | Affects isolation efficiency and expansion |
| Prior treatments | T cell viability, phenotype | Impacts activation and transduction efficiency |
| Collection protocol | Cell composition, purity, and viability | Influences process consistency |
| Storage and transport conditions | T cell count, phenotype distribution, and functional capacity | Reduces achievable dose |
Selecting the right isolation strategy can help buffer that variability. Commonly used approaches include:
- Fluorescence-activated cell sorting (FACS): A flow cytometry-based technique best for small scales in discovery and R&D, offering ultra-high purity isolation with multi-marker precision.
- Magnetic-activated cell sorting (MACS): Magnetic beads enable very gentle selection best for large volumes and high-throughput processing while also offering compatibility with closed, GMP-friendly systems. Using antibodies or ligands on the surface of the beads, cells can be sorted using positive or negative selection:
- Positive selection: Target cells bind to the bead bind surface. Preferred for high purity and specificity but requires additional processing.
- Negative selection: Unwanted cells bind to the bead surface, leaving target cells untouched. Preferred to maintain viability and performance of target cells, but purity may be lower. Microbubble-based negative selection may be able to offer high purities with the benefits of a simpler workflow.
- PBMC enrichment: A density gradient centrifugation procedure sometimes used when T cell isolation is not required, or the product has high red blood cell contamination.
Because the quality of isolated T cells directly influences therapeutic success, many developers are moving away from manual isolation workflows that introduce operator-derived variability in timing, bead exposure, wash efficiency, and handling stress—all of which can amplify differences already present in patient starting material.
Process considerations for scalable, GMP‑ready T cell isolation
In early development, it’s essential to build isolation protocols that can accommodate highly varied patient material and be amenable to scale. Otherwise, you may be faced with costly process changes later in development. For autologous programs, scaling centers around lot-to-lot consistency, rather than producing large, single batches.
Automated, closed-system approaches help standardize critical parameters and provide more consistent T cell recovery and viability across highly variable inputs. This consistency becomes especially important as programs move toward clinical manufacturing, where reproducibility and documentation are as critical as yield.
Read our application note on automated isolation in CAR T manufacturing.
Activation and gene transfer in the CAR T manufacturing process
Once T cells are isolated, the next step is to activate them and introduce the genetic material responsible for chimeric antigen receptor (CAR) expression, which enables T cells to recognize and destroy cancer cells. This phase is central to transforming a patient’s own immune cells into their personalized cancer therapy.
T cell activation: preparing cells for gene transfer
Activation typically involves stimulating the T cells using antibodies (e.g., anti-CD3/CD28) or cytokines like interleukin-2 (IL-2) to mimic signals they would receive during an immune response. Proper activation primes the cells for efficient gene transfer and proliferation.
While activation strategies are relatively consistent across CAR T programs, the choice of gene transfer method has significant implications for scalability, regulatory complexity, and long‑term manufacturing strategy.
Gene transfer strategies
Gene transfer immediately follows activation and is most commonly achieved using viral vectors, which provide stable integration of the CAR gene into the T cells for long-term expression.
All approved CAR T therapies use either lentiviral or gamma-retroviral vectors for gene delivery, providing strong regulatory precedent for both options. Let’s take a closer look at these systems:
| Lentiviral vectors | Retroviral vectors | |
|---|---|---|
| Cell activation state | Dividing and non-diving | Dividing only |
| Workflow impact | Tolerates a range of T cell activation strength | Dependence on T cell activation state; narrow process window |
| Advantages |
|
|
| Challenges |
|
|
In practice, the choice often reflects a balance between biological requirements, manufacturing scalability, and available vector supply infrastructure, rather than transduction efficiency alone.
When should teams consider non‑viral gene delivery?
Non-viral delivery methods, including electroporation (EP), are most often used for transient payloads, rather than for delivering CAR constructs intended for long‑term expression. These approaches can reduce reliance on viral vector supply but are more prone to variable outcomes (due to cell stress) and are more difficult to scale.
As a result, transient gene expression is most suitable for discovery, genome‑editing workflows (e.g., CRISPR‑Cas9 reagents), or next‑generation CAR designs that don’t require persistence within a patient.
Lipid nanoparticles (LNPs) are an emerging alternative for CAR mRNA delivery, offering a gentler, more manufacturing-friendly approach compared to EP. As delivery efficiency and persistence improve, LNP‑based strategies may play a larger role in future CAR T platforms. Read more about the growing role of LNPs in cell therapy.
Cell expansion strategies for CAR T therapy manufacturing
Cell expansion follows gene transfer, allowing the modified T cells to multiply to clinically relevant doses within nutrient-rich expansion media, typically supplemented with serums and growth factors. Although the total number of required cells varies by therapy design and indication, all CAR T workflows depend on robust, scalable expansion.
This phase is a major determinant of whether developers can consistently achieve target doses. T cells need specific physio-chemical conditions to expand effectively, and these parameters must be carefully optimized during process development to support viability and growth. Key factors include:
- Environmental: temperature, pH, O₂/CO₂
- Nutritional: nutrient availability, media composition
- Mechanical: shear stress
Small deviations in these parameters can have outsized effects on viability and expansion kinetics.
Early CAR T workflows often use static bioreactors such as G‑Rex flasks, which provide adequate surface area and gas exchange for small‑batch expansion. However, as processes scale, gas transfer, nutrient gradients, and mixing limitations become more pronounced. For this reason, developers frequently transition from static cultures to fed-batch expansion in rocking bioreactors or automated systems.
In a fed‑batch process, fresh nutrients are added throughout the expansion protocol rather than only at the start. Gentle mixing helps maintain uniform nutrient distribution, supporting more prolific and consistent CAR T expansion at clinical scale.
Learn more about bioprocessing fundamentals: Bioreactor types and modes of operation
Future‑proofing cell expansion for the clinic and commercialization
Teams beginning in static systems should assess early how easily their protocol can transition to more controlled, GMP-friendly expansion environments. In addition to hardware considerations, the choice of media and reagents used for expansion becomes increasingly important as programs advance.

One example is the industry-wide shift to serum-free media, which helps reduce variability associated with animal‑derived components (such as fetal bovine serum), simplify regulatory compliance, and improve product quality. However, changing serums often requires re-optimization of growth conditions and can affect expansion kinetics.
It’s never too soon to familiarize yourself with regulatory guidance and GMP considerations. Making the right decisions early can set you up for a smooth transition to commercialization.
Read our application note on automated CAR T cell expansion in serum-free media.
Harvest and formulation of CAR T cells
After expansion, CAR T cells must be harvested, washed, and formulated into a final product suitable for patients. Because the expanded cells represent the full therapeutic dose, this stage plays a critical role in preserving viability and maintaining product consistency.
Harvesting expanded CAR T cells
Cultured cells are collected from bioreactors or culture bags, followed by washing to remove residual media, reagents, and cellular debris. Then cells are typically concentrated and resuspended in an appropriate buffer (or cryoprotectant) for formulation.
Why harvest and washing are high-risk steps: The core challenge is achieving high cell recovery without compromising viability. Abrupt osmotic shifts or excessive shear stress due to repeated centrifugation can reduce viability, while overly gentle processing can result in high cell loss.
Centrifugation-based workflows used in early development scale poorly. Multiple spin–resuspend cycles increase handling time, operator variability, and cumulative cell loss, and they become increasingly difficult to standardize under GMP conditions.
Mitigating risks through precision: With larger scales, automated systems are increasingly used to control wash conditions more precisely. These platforms can regulate flow rates, buffer exchange, and processing time to minimize mechanical stress while maximizing recovery, helping developers achieve more consistent yields across batches.
Formulating the CAR T therapy dose
To prepare the product for infusion or cryopreservation, cells are often divided across multiple bags depending on required number of doses. Each dose should contain the target cell count, with allowances for anticipated cell loss after cryopreservation.
At this stage, consistency and safety depend on precise handling and robust sterility controls. Closed, automated systems that employ single-use consumables support these requirements by eliminating open handling, reducing contamination and batch failure risk.
Read our application note on closed, automated CAR T cell harvest and formulation.
How to gain regulatory readiness
Digital data capture and electronic SOPs embedded in many automated systems also simplify sterility assurance and batch documentation. Once a nice-to-have, these capabilities are now foundational to process reliability, productivity, and regulatory readiness as programs advance toward clinical manufacturing.
Cryopreservation of CAR T cells
Cryopreservation serves as a critical step to preserve the quality and stability of the final CAR T product during storage and transport prior to clinical use. Because CAR T therapies are patient‑specific, failure at this stage often leaves no backup material.
Cells are highly sensitive to temperature and cooling rate: too rapid or too slow cooling can dramatically reduce viability. Any loss of viable cells at this point results in a lower delivered dose, which may compromise therapeutic effectiveness.
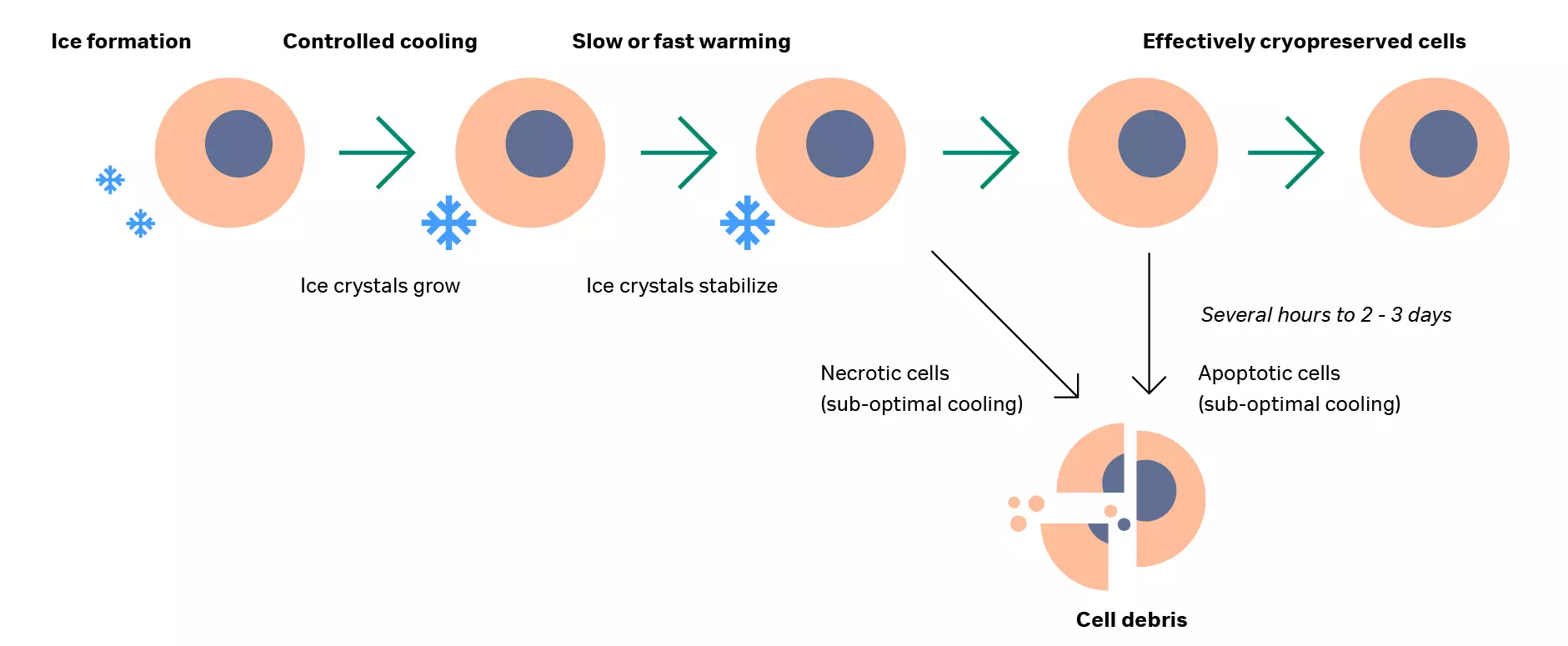
Early workflows often rely on liquid nitrogen, although not ideal for commercialized cell therapy products given liquid nitrogen can become contaminated with particles and biologically active bacteria.
Conduction-based cryopreservation methods offer a controlled alternative, enabling precise management of temperature and cooling rates with comparable outcomes to traditional liquid nitrogen methods. These systems also introduce other advantages, including improved safety, simpler operation, and reduced energy consumption.
Learn more about techniques to maximize T cell viability.
Thawing cryopreserved CAR T cells at the point of care
Thawing of the cryopreserved product is typically performed at the point of care, shortly before the CAR T therapy gets administered to the patient. As with cryopreservation, improper thawing, such as rapid jumps in temperature, can compromise cell viability and reduce therapeutic effectiveness.
A traditional thawing process involves submerging a frozen sample in a water bath at 37°C and visually determining when the last ice has melted. Although inexpensive and suitable for some research settings, water bath thawing produces poses contamination risks and often produces inconsistent results. Variability largely stems from uncontrolled conditions and variation in operator-dependent technique; even clinical staff may struggle to achieve consistent outcomes with manual thawing.
Because thawing happens outside the manufacturing facility, developers must ensure the process is simple and reproducible across clinical sites.
Dry thawers can execute automated thawing procedures with precise temperature control, minimal contamination risk, and reproducibility across sites while delivering comparable performance to water bath methods.
Learn more about how cells handle temperature changes in our guide to cell thawing.
Frequently asked questions
1. What changes when a CAR T process moves from bench-scale research to GMP manufacturing?
Practically, teams often need to transition open and manual steps to closed or functionally closed workflows, tighten acceptable process parameters, and introduce in-process controls that support documentation for lot release.
GMP manufacturing requires controlled materials, documented SOPs and batch records, qualified facilities and equipment, validated processes and analytics, and a robust quality system.
2. Which unit operations are usually the biggest scalability bottlenecks in autologous CAR T manufacturing?
Bottlenecks often come from sources of variability: apheresis starting material, manual T cell enrichment and isolation procedures, expansion parameter control, and cell loss in downstream processing. These steps drive batch-to-batch variability, making it difficult for teams to standardize and scale out.
3. What are common contamination risks in CAR T manufacturing, and what prevention strategies are most effective?
Contamination risk is highest during open handling but can also come from connector or assembly failures and poorly controlled starting material. The most effective prevention is designing a closed or functionally closed workflow, minimizing manual interventions, standardizing sterile connection methods, and maintaining robust training and environmental monitoring.
4. How do developers handle process changes (e.g., switching media or moving to a closed system) without jeopardizing clinical comparability?
The key is establishing a baseline process and analytics package before you change anything. A change control strategy should include a clear rationale, risk assessment, and comparability plan. Depending on the change and development stage, this can include side-by-side runs, bridging studies, and statistical evaluation of key outputs (e.g., phenotype distributions, viability, vector copy number, potency).
Designing your process with future transitions in mind can reduce the comparability burden (and potential complications) later.
5. What are the most common points of failure outside the manufacturing facility (logistics, cryopreservation, and thawing), and how can teams reduce risk?
Autologous therapies are uniquely vulnerable to chain-of-identity and chain-of-custody risks. Common failure modes include inconsistent apheresis handling, inadequate shipping conditions, uncontrolled freeze or thaw rates, long hold times, and site-to-site variability in thawing technique.
Teams can reduce these risks by standardizing collection procedures, using qualified shippers and shipping lanes, validating cryopreservation and thaw workflows, and providing simple, reproducible instructions for point-of-care handling supported by training and automated equipment.
6. How do you decide whether to manufacture in-house vs. use a CDMO for early clinical batches?
Choose based on speed, capabilities, and control. A CDMO can shorten timelines if they already have the space, trained staff, and quality systems, but capacity queues and tech transfer can slow you down. In-house manufacturing offers tighter control over scheduling and process learning, but requires major investment in facilities, quality systems, and staffing. Many teams start with a CDMO for Phase 1, then bring it in-house as the process stabilizes.
Dive deeper: Step up to GMP manufacturing or outsource to a CDMO?