As the global pharmaceutical industry approaches a key moment—the so-called ‘biosimilar patent cliff’—the opportunity for biosimilar development has never been greater. For emerging biotech companies and CDMOs, this shift offers both a challenge and a chance to redefine access to therapies. At Cytiva, our regulatory support and services teams are available to help companies chart their course with confidence.
The patent cliff and the race to market
More than $200 billion in annual biologic drug revenue is at stake as more than a dozen patents expire over the next five years. For smaller manufacturers, biosimilars offer a lower-cost path to market – often 15 to 20 times less expensive in R&D than innovator biologics (1). But the race is fierce.
Missing a regulatory deadline can mean losing first-mover advantage, which could translate into millions of lost revenue. Sandra Racordon-Pape, Director Regulatory Science and Strategy at Cytiva, shares a real-world example: “One company lost its competitive edge because several of its competitors launched ahead of them while they were still resolving regulatory issues.”
It goes without saying that speed is critical in this competitive environment. To that end, navigating the regulatory landscape should be top of mind when developing a biosimilar. From demonstrating biosimilarity to managing good manufacturing practices (GMP) compliance and documentation, the path to market is fraught with potential delays.
What’s driving regulatory complexities
Despite the promise of biosimilars to reduce healthcare costs and expand access to biologics, their regulatory pathways are much more involved than those for traditional generics. Several factors are at play (2, 3):
- Scientific and manufacturing complexity: Biosimilars are large, structurally intricate molecules that come from living cells. This inherent variability means they can never be exact copies of their reference products. Regulatory agencies require extensive analytical, nonclinical, and clinical data to show ‘high similarity’ that doesn’t bring clinically meaningful differences in safety, purity, or potency.
- Divergent global regulatory frameworks: While agencies like the US Food and Drug Administration (FDA), European Medicines Agency (EMA), and World Health Organization (WHO) have established biosimilar guidelines, these frameworks aren’t harmonized (see Table 1). Developers must tailor regulatory strategies for each target market, which increases both cost and time to approval. When a biosimilar is destined for a global market, it’s crucial to understand the nuances of getting approval from all the relevant regulatory agencies.
- Evolving guidelines and expectations: Regulatory agencies periodically update their guidance to reflect advances in analytical technology, real-world evidence, and post-marketing surveillance. Developers must stay on top of these changes and adapt their programs to align with these changes.
- Interchangeability and substitution barriers: Achieving interchangeability status in the US requires additional clinical switching studies, where patients alternate between the reference product and the interchangeable biosimilar. There must be no decrease in effectiveness and no increase in safety risk associated with switching. In Europe, substitution isn’t always permitted; this can slow down adoption of the approved therapy.
- Patent and exclusivity challenges: Even after regulatory approval, biosimilars often face legal challenges from originator companies. Among these are patent thickets, exclusivity extensions, and litigation that can delay market entry.
Taken together, these factors explain why biosimilar development isn’t a ‘plug-and-play’ process. It requires a strategic approach.
Table 1. Comparing global regulatory frameworks for biosimilars: FDA, EMA, and WHO
| Feature | FDA (US) (3) | EMA (Europe) (4) | WHO (global guidance) (5) |
| Definition | Highly similar to reference product with no clinically meaningful differences | Similar biological medicine to a reference product in terms of quality, safety, and efficacy | Similar biotherapeutic product to a licensed reference product |
| Approval pathway | IND-BLA | CTA-MAA | The national regulatory authority (NRA) is responsible for clearly defining a suitable regulatory framework for licensing biological products, including biosimilars |
| Major legislation, directives, and regulations | • Biologics Control Act (safety and efficacy) • Biologics Price Competition and Innovation Act of 2009 (BPCIA) • 21 CFR: o 210, 211 (cGMP-drug) o 312 (IND) o 351(k) (BLA abbreviated licensure pathway) |
• Directive 2001/83/EC • Directive 2017/1572 (cGMP) • Regulation 726/2004/EC (MAA) • Regulation 1252/2014 (cGMP for API) • Regulation 536/2014 (Clinical trial) |
WHO Guidelines on Evaluation of Biosimilars (2022) Guidelines may be adopted as definitive national requirements, or modifications may be justified and made by the NRA |
| Key requirements | • Analytical, nonclinical, and clinical studies • Interchangeability pathway available • Risk management plan |
• Stepwise approach: quality, nonclinical, clinical comparability • Risk management plan |
Emphasizes stepwise comparability; supports regulatory convergence |
| Interchangeability | Separate designation; allows pharmacy-level substitution | No formal designation; substitution policies vary by country | Not addressed directly; left to national authorities |
| Scientific advice | BIA meeting | Scientific advice and protocol assistance available | Encourages early engagement with national regulators |
IND is Investigational New Drug application. BLA is Biologics License Application. CTA is Clinical Trial Application. MAA is Marketing Authorization Application. cGMP is current good manufacturing practices. BIA is Biosimilar Initial Advisory meeting. API is active pharmaceutical ingredient.
Regulatory documentation: the backbone of biosimilar success
Regulatory documentation isn’t just a compliance requirement – it’s a strategic asset. Our regulatory support and services are designed to help biosimilar developers build, manage, and submit the right documentation at the right time to the right authorities.
Documentation support includes:
- Validation guides: Help Cytiva customers validate systems and processes.
- Validation support files (VSF): Provide test data and traceability for compliance.
- Regulatory support files (RSF): Ensure alignment with agency expectations.
- Change control notifications (CCN): Keep customers informed of updates that may impact filings.
These documents are essential for maintaining compliance and ensuring that submissions are complete, accurate, and tailored to the expectations of each regulatory body. Underscoring this point, Peter Trebicki, Director, Regulatory Customer Care at Cytiva, says, “Even though formats are similar, EMA, FDA, and National Medical Products Administration (NMPA) in China, for example, each have unique requirements. Missing a detail can mean missing a deadline.”
Regulatory services: meetings, submissions, and so much more
Engaging with regulatory authorities can be overwhelming. Our team helps biosimilar developers prepare for regulatory meetings by building briefing packages and advising on how to engage with agencies. As Sandra explains, “We help derisk applications by guiding customers through early interactions with regulators, which can clarify expectations and prevent missteps.”
Our team supports the drafting of the chemistry, manufacturing, and controls (CMC) sections of regulatory submissions. This includes translating technical data into regulatory language and ensuring that all manufacturing processes are clearly documented and aligned with the reference product.
Another service is performing a biosimilar review, acting as a neutral third party to assess whether the approach to demonstrating biosimilarity is sound. If data has already been generated, our team can review it and advise on how to demonstrate equivalence to reference products. This is an area where many developers need help. Sandra says, “We've seen some rejections where they haven’t been able to prove the biosimilarities sufficiently.”
Unequivocal approval by regulatory authorities is the goal. But that doesn’t always happen. We can help address complete response letters (CRLs) and European public assessment report (EPARs), both of which are highly specific and have a tight timeline for resolution. What sorts of major deficiencies have other companies received? One example is comparability between the proposed biosimilar and its originator biological. Another is when regulatory bodies highlighted process validation as a major issue as well as cGMP related issues, like deficiencies in process controls, equipment sterilization, and vial filling strategies.
Supporting startups and CDMOs
Our regulatory support and services are especially valuable for startups and CDMOs. Startups often lack in-house regulatory expertise. Our team can help them build roadmaps, understand territory-specific requirements, and prepare for clinical trials. “We guide them through which regulations apply to which markets and when to submit what documentation,” says Sandra.
For CDMOs, the focus is often on GMP compliance. For example, Cytiva has previously supported a CDMO in preparing for inspections and resolving environmental monitoring issues in the aseptic filling step. Even established CDMOs sometimes need help. We’re here to support both the manufacturer and their partners. Figure 1 highlights our documentation, know-how, and experience.
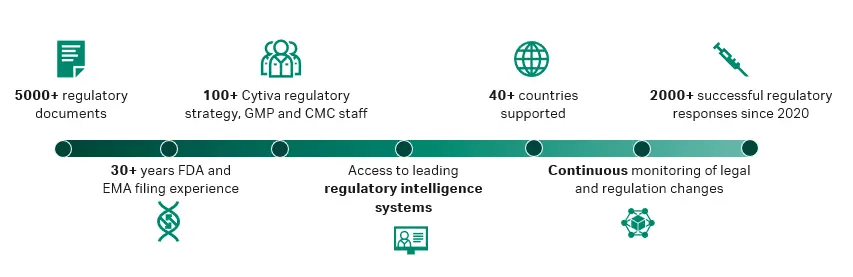
Fig 1. Why choose Cytiva for regulatory support and services?
Final advice: be proactive, be prepared
Sandra and Peter offer three key takeaways:
- Know your partners: If you’re working with a CDMO, vet them carefully and ensure they’re GMP compliant.
- Engage early with regulators: Early feedback can save time and money.
- Keep documentation current: “It’s not just about compliance – it’s about speed,” says Peter.
With the right documentation, regulatory strategy, and support, biosimilar developers are well on their way to not only meet the necessary regulatory standards but also accelerate their path to market – and to patients.
REFERENCES
- Lechleider T. American Century Investments. Biosimilars: A cure for the higher price of complex drugs? Published March 11, 2024. Accessed July 15, 2025. https://www.americancentury.com/insights/biosimilars-cure-for-the-higher-price-of-complex-drugs/
- Monga A, Gagan, Jamwal P, Sharma S, Kaur A. Biosimilars: A critical review of development, regulatory landscape, and clinical implications. AAPS PharmSciTech. 2025;26:46. https://doi.org/10.1208/s12249-025-03038-2.
- DrugPatentWatch. Updated June 24, 2025. Accessed July 15, 2025. https://www.drugpatentwatch.com/blog/top-5-challenges-faced-biosimilars/
- US Food and Drug Administration. Published March 1, 2023. Accessed July 15, 2025. https://www.fda.gov/drugs/therapeutic-biologics-applications-bla/biosimilars
- European Medicines Agency. Accessed July 15, 2025. https://www.ema.europa.eu/en/human-regulatory-overview/biosimilar-medicines-overview
- World Health Organization. Guidelines on evaluation of biosimilars. Published April 22, 2022. Accessed July 15, 2025. https://www.who.int/publications/m/item/guidelines-on-evaluation-of-biosimilars