Summary
The biopharmaceutical industry continues to incorporate single-use technology to improve flexibility, and plant efficiency. A single-use manufacturing process from start to bulk fill may include hundreds of polymeric consumables. Extractables data are provided by the suppliers but the responsibility is on the drug product manufacturer to assess these extractables data for their specific process and generate leachables data where needed to ensure a safe and compliant product. A leachables study can be time consuming and costly, hence it is crucial to optimize any leachables testing.
This paper describes a step-by-step procedure on how to use Cytiva extractables data and current industry best practices for leachables risk assessment of a complete single-use generic monoclonal antibody (mAb) manufacturing process. Such assessment will help to determine where there are risks that may require additional studies. This will lead to better understanding of patient risk as a result of the use of single use consumables in the drug manufacturing process under normal processing conditions.
Our assessment found that all the consumables in the process, ending with bulk fill, were associated with low or medium risk. Requirements on compendial testing and extractables data were fulfilled according to industry best practices. A step-by-step procedure was used to calculate extractables per surface area of consumables and obtain an extractables data set representative of potential accumulated leachables in the final bulk fill bag. A chemical safety assessment performed on the results indicated that the potential leachables generated during the process ending with the bulk fill, would be below levels of concern. Hence, the safety risk was found acceptable within the assumptions discussed and did not imply that a leachables study would be needed prior to final fill and finish.
Single-use consumables in biomanufacturing processes
Adoption of single-use technologies, i.e., gamma-sterilized plastic components that are used once and discarded by biopharmaceutical manufacturers, is a key element in reducing manufacturing costs, improving plant flexibility and turnaround times, as well as producing safe and regulatory compliant products. Single-use technologies (SUTs) are used throughout the biopharmaceutical manufacturing processes.
Large number of different polymeric consumables are used throughout a single-use process. More than 300 items are typically included in the consumables bill of material (BOM) for a 2000 L mAb process using Cytiva single-use manufacturing equipment, outlined in Figure 1. The consumables BOM encompasses bioreactor bags of different sizes, mixer and hold bags, filters, transfer tubing, chromatography columns, chromatography and filtration flow kits, and bags for holding the bulk drug substance (BDS).
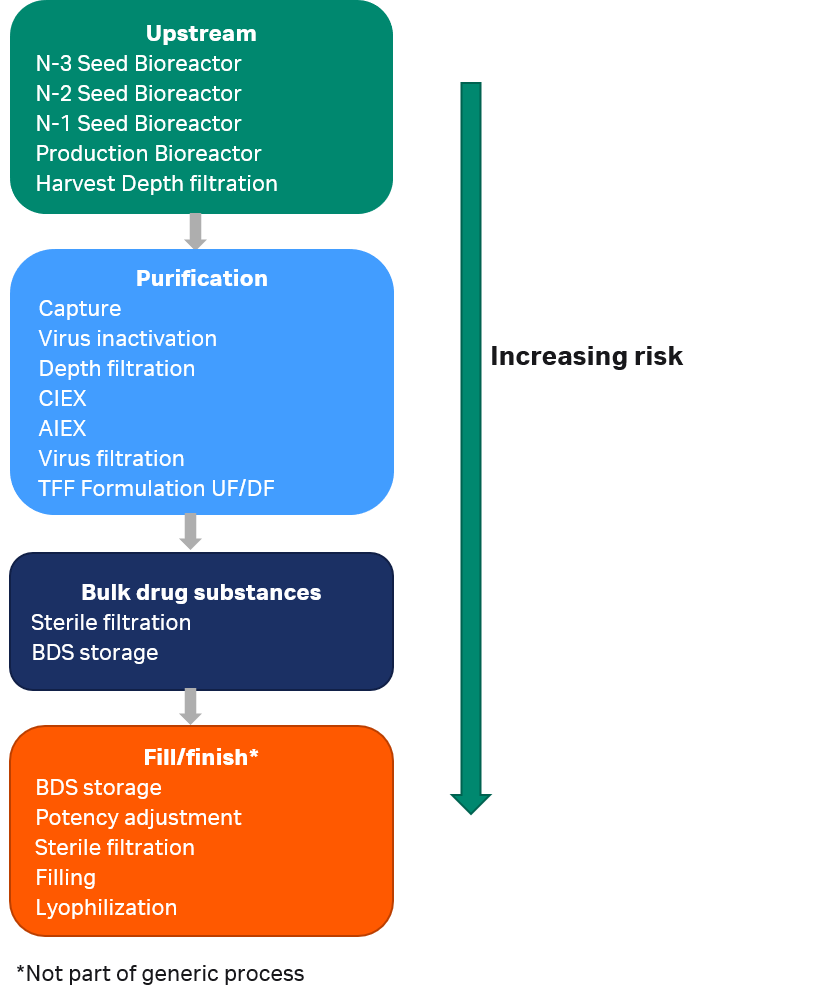
Fig 1. Process steps for a mAb manufacturing process. Patient risk related to leachables increases with distance along the production stream.
Extractables data
Extractables screening is crucial to provide a profile of potential substances that may be extracted into the process stream. Regulatory bodies require that any production equipment used in biopharmaceutical manufacturing must not present any hazard to the final drug product (1).
Extractables data are by definition generated at exaggerated conditions relative to the actual process conditions. In the standardized extractables protocol recommended by the BioPhorum (2) and which Cytiva adheres to, pre-defined model solvents are used at increased temperature and several contact duration timepoints to exceed anticipated use conditions of the consumable, followed by extensive chemical analysis, as summarized in Figure 2.

Fig 2. BioPhorum recommendations for performing an extractables study.
Cytiva has a long history of performing extractables studies on single-use consumables starting from the introduction of ReadyToProcess products in 2007. Study designs have evolved over time; the selection of solvents have become broader, exposure duration longer, and analytical methods more sophisticated. Studies have been performed on complete assemblies (e.g., ÄKTA ready Flow kit), components (e.g., ReadyMate disposable aseptic connector, multilayer bag films), and separate materials (e.g. polyethylene material of bag ports). Also, the push on component manufacturers to provide extractables data to integrators of completed assemblies, like Cytiva, has resulted in new published studies now available for sensors, connectors, tubing, etc. Thus, extensive amounts of extractables data, partly overlapping between assemblies and separate components/materials are now available. These data are provided to Cytiva customers in Validation guides and separate Extractables reports available on the Cytiva Regulatory support web page.
How to use industry best practices for evaluating leachables risk in your manufacturing process
Extractables data are valuable if they allow for a reasonable prediction of potential leachables. Extractables data can be used as basis to perform toxicity assessments, to plan a leachables study, or to provide reasons to avoid a detailed leachables study. The expenses for leachables testing in terms of cost and time can become large and therefore the focus should be at the right phase of the biomanufacturing process and on the right consumables.
The BioPhorum Best practices guide for evaluating leachables risk from polymeric single-use systems in biopharmaceutical manufacturing, provides guidance for how to perform a risk assessment of your process and decide what leachables testing is necessary (3).
In this paper, we are using the BioPhorum guidance and available extractables data from Cytiva to assess the risk with leachables from single-use consumables in a generic mAb manufacturing process (outlined in Fig 1). The different steps of this evaluation are described in Figure 3.

Fig 3. Step-by-step evaluation to assess leachables risk.
Extractables and Leachables propensity assessment
When the complete bill of materials (BOM) is available for all process consumables, a first step is to perform a propensity assessment to rate risk, depending on where in the process and under what conditions the consumable is used. If the material is not polymeric or the polymeric material is not in contact with process fluid, such components do not need further evaluation and can be removed from the risk assessment. For all polymeric components in contact with process fluid, the point of use in the process will heavily weigh the risk for leachables.
In addition to the location of the consumable along the production stream, actual process conditions need to be considered. Temperature, contact time, type of process fluid, and wetted surface area will affect the propensity for leachables as described below.
- Temperature — higher temperature increases the possibility of compounds to migrate into surrounding fluid from a material.
- Duration — exposure duration needs to be considered because the length of exposure may increase the propensity for compounds migrating into surrounding fluid from a material.
- Solvent interaction — poor chemical compatibility of a polymeric material with a process fluid will lead to increased levels of leachables.
- Surface area to volume ratio — higher surface areas provide more opportunity of leaching. Lower process volumes concentrate the leachates and may represent higher risk.
For our assessment, the risk categorization model suggested in the BioPhorum leachables guide was used without modifications, so that the progress along the production stream and process conditions were weighted and rated as summarized in Table 1. Surface areas of wetted components were added to the BOM together with information on process volume in contact with the consumable. The process conditions for temperature, exposure duration, and type of process fluid were estimated and categorized according to the model in Table 1.
Table 1. Leachables risk assessment model (3).
| Consideration | Weight | Rating | Comment |
|---|---|---|---|
| Progress along the production stream | 0.4 | 1 | Upstream e.g., working cell bank, vial thaw, inoculum, expansion, production, harvest, plasma, solution preparation |
| 3 | Purification e.g., filtration, chromatography, viral inactivation, viral filtration, UF/DF |
||
| 5 | Bulk drug substance e.g., formulation, 0.22 μm filtration, BDS storage |
||
| 9 | Final formulation, fill/finish, e.g., bulk drug product storage, potency adjustment, sterile filtration, filling |
||
| Exposure temperature | 0.15 | 1 | < 0˚C |
| 3 | 0 to 8˚C | ||
| 5 | > 8 to 30˚C | ||
| 9 | > 30˚C | ||
| Exposure duration | 0.15 | 1 | Transient (≤ 60 minutes) |
| 3 | Short (≤ 24 hours) | ||
| 5 | Medium (≤ 7 days) | ||
| 9 | Long (> 1 week) | ||
| Process fluid interaction | 0.15 | 1 | Limited penetration into polymeric component, e.g., water |
| 3 | Low solvation power or low penetration of polymeric component, e.g., neutral pH without organics, surfactants, etc. | ||
| 5 | Medium solvation power or medium penetration of polymeric component, e.g., surfactant, low-concentration organics, high/low pH solutions without organics/detergents | ||
| 9 | High solvation power or high penetration of polymeric component | ||
| Dilution ratio (Surface area to volume ratio) | 0.15 | 1 | < 1 × 10-3 m2/L (0.01 cm2/mL) |
| 3 | 1 × 10-2 m2/L to 1 × 10-03 m2/L (0.01 to 0.1 cm2/mL) | ||
| 5 | 1 × 10-1 m2/L to 1 × 10-02 m2/L (0.1 to 1.0 cm2/mL) | ||
| 9 | > 1 × 10-1 m2L (> 1 cm2/mL) |
Risk categorization
Based on the model, risk scores will theoretically range from 1 to 9, and be classified at three levels: Low for risk score 1.0 to 3.6, Medium for risk score 3.7 to 6.2, and High for risk score 6.3 to 9.0. Depending on risk classification, different requirements have been suggested for components of polymeric materials, as summarized in Table 2.
Table 2. Requirements based on risk classification of polymeric components.
| Risk score | Risk classification | Suggested requirement |
|---|---|---|
| 1.0 to 3.6 | Low | Meets compendial requirements (e.g., USP, EP, etc.) |
| 3.7 to 6.2 | Medium | Low-risk requirements, plus extractables data evaluation that brackets the intended use available (per BioPhorum testing recommendation) |
| 6.3 to 9.0 | High | Medium-risk requirements, plus satisfactory extractables data evaluation that brackets the intended use and relevant E and/or L profile |
Examples from the outcome of risk classification performed on three bags used at different locations in the process are shown in Table 3.
Table 3. Examples of leachables risk rating of bags used in upstream, purification and bulk drug substance.
| Parameter: | Distance along process stream | Exposure temperature | Exposure duration | Solvation power and penetration | Dilution factor | Risk score (1 to 9) |
Risk classification |
|---|---|---|---|---|---|---|---|
| Weight: | 0.40 | 0.15 | 0.15 | 0.15 | 0.15 | ||
| Upstream, 100 L bag for buffer preparation | |||||||
| Bag film | 1 | 5 | 9 | 5 | 5 | 4.0 | Medium |
| Impeller | 1 | 5 | 9 | 5 | 3 | 3.7 | Medium |
| Tubing | 1 | 5 | 9 | 5 | 3 | 3.7 | Medium |
| Ports | 1 | 5 | 9 | 5 | 1 | 3.4 | Low |
| Connectors | 1 | 5 | 9 | 5 | 1 | 3.4 | Low |
| Plugs | 1 | 5 | 9 | 5 | 1 | 3.4 | Low |
| Swab-able valve | 1 | 5 | 9 | 5 | 1 | 3.4 | Low |
| Purification, 2500 L bag for product hold after virus filtration | |||||||
| Bag film | 3 | 5 | 3 | 3 | 5 | 3.6 | Low |
| Impeller | 3 | 5 | 3 | 3 | 1 | 3.0 | Low |
| Tubing | 3 | 5 | 3 | 3 | 1 | 3.0 | Low |
| Ports | 3 | 5 | 3 | 3 | 1 | 3.0 | Low |
| Connectors | 3 | 5 | 3 | 3 | 1 | 3.0 | Low |
| Plugs | 3 | 5 | 3 | 3 | 1 | 3.0 | Low |
| Swab-able valve | 3 | 5 | 3 | 3 | 1 | 3.0 | Low |
| Bulk fill, 100 L storage bag holding BDS after sterile filtration | |||||||
| Bag film | 5 | 3 | 9 | 3 | 5 | 5.0 | Medium |
| Tubing | 5 | 3 | 9 | 3 | 3 | 4.7 | Medium |
| Ports | 5 | 3 | 9 | 3 | 1 | 4.4 | Medium |
| Connectors | 5 | 3 | 9 | 3 | 1 | 4.4 | Medium |
| Plugs | 5 | 3 | 9 | 3 | 1 | 4.4 | Medium |
| Swab-able valve | 5 | 3 | 9 | 3 | 1 | 4.4 | Medium |
All components of consumables in the generic mAb manufacturing process were categorized with low or medium risk, as illustrated in Figure 4. None were categorized as high risk, i.e., found with a risk score ≥ 6.3. Fulfilment of relevant compendial testing such as biocompatibility testing is sufficient for a component classified with low risk. All Cytiva consumables fulfill such compendial testing requirements. Components categorized as medium risk are, in addition to compendial testing requirements, also required to carry supportive extractables data that brackets the intended use. These components were considered in the next step for risk mitigation.
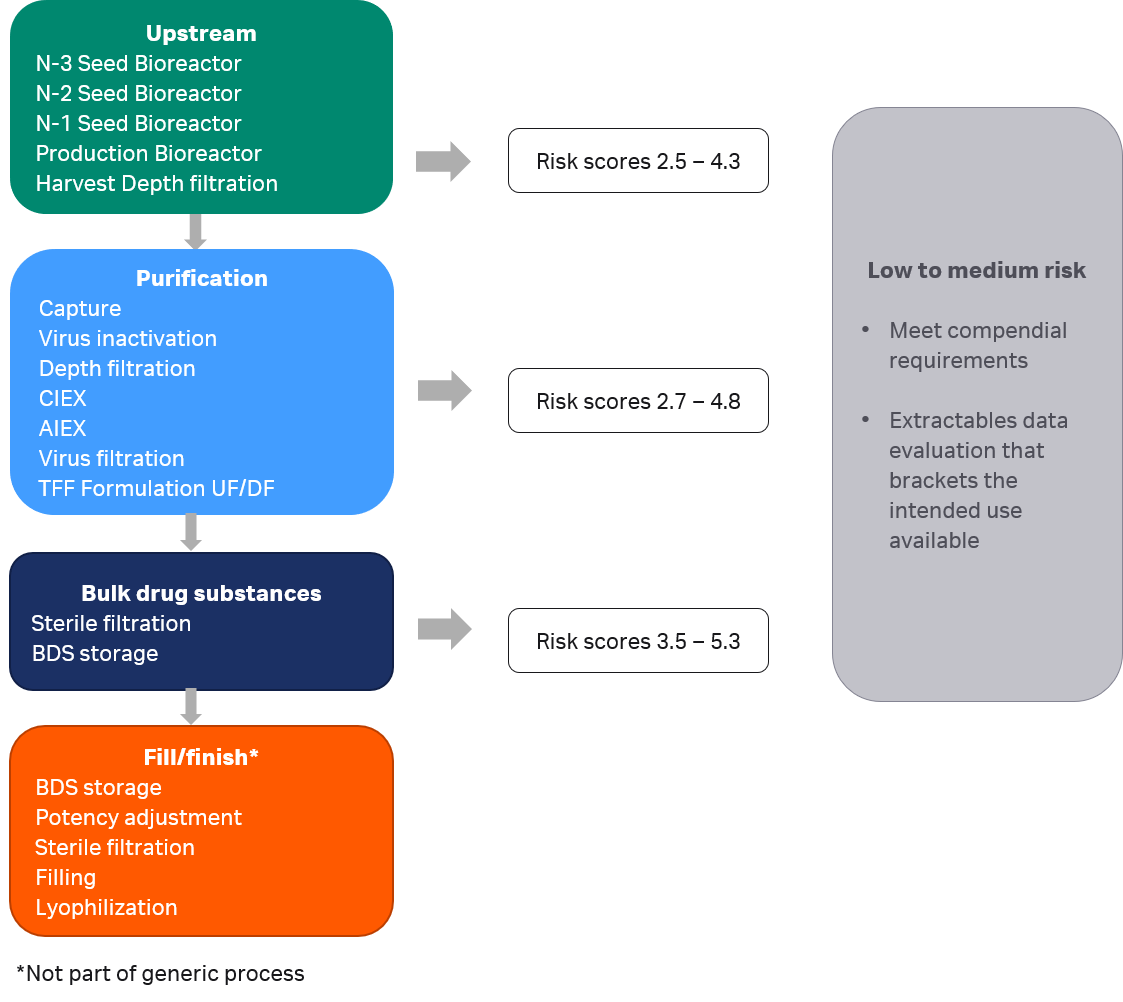
Fig 4. Risk scores for components of consumables in the generic mAb manufacturing process.
Risk mitigation

Once a risk assessment has been performed certain aspects of the manufacturing operation should be considered when making a risk-based decision on whether to proceed with a leachables study.
It is generally accepted that certain process steps will reduce leachables to a large extent, i.e., behave as sinks of leachables. In the upstream process leachables from a bioreactor are known to be removed during harvest as these are adsorbed on host cell surfaces and cell debris (4). During downstream purification, membrane filters can contain substance specific scavenger capacities in the range of µg/cm2 of nominal membrane surface (5). Also, chromatography resins or membrane adsorbers have typical substance specific scavenger capacities in the range of µg/cm2 of membrane adsorbent bed volume.
The largest reduction in leachables usually occurs in formulation steps. Ultrafiltration is commonly used in the bioprocess for concentrating a dilute product stream whereas diafiltration is used to buffer exchange the product into the desired buffer system, both are effective to remove small molecules such as extractables and leachables (6,7). Various studies have demonstrated a 100 to 10,000-fold reduction of small molecule concentrations from load to final filtrate in standard diafiltration processes (8). Also, efficient clearance of extractables and leachable compounds has been demonstrated during filtration processes in the presence of proteins (9).
When the likelihood of removing leachables in process steps has been considered, consumables used in unit operations before a UF/DF filtration step may be assigned to a lower risk classification and removed from further assessment as illustrated in Figure 5.
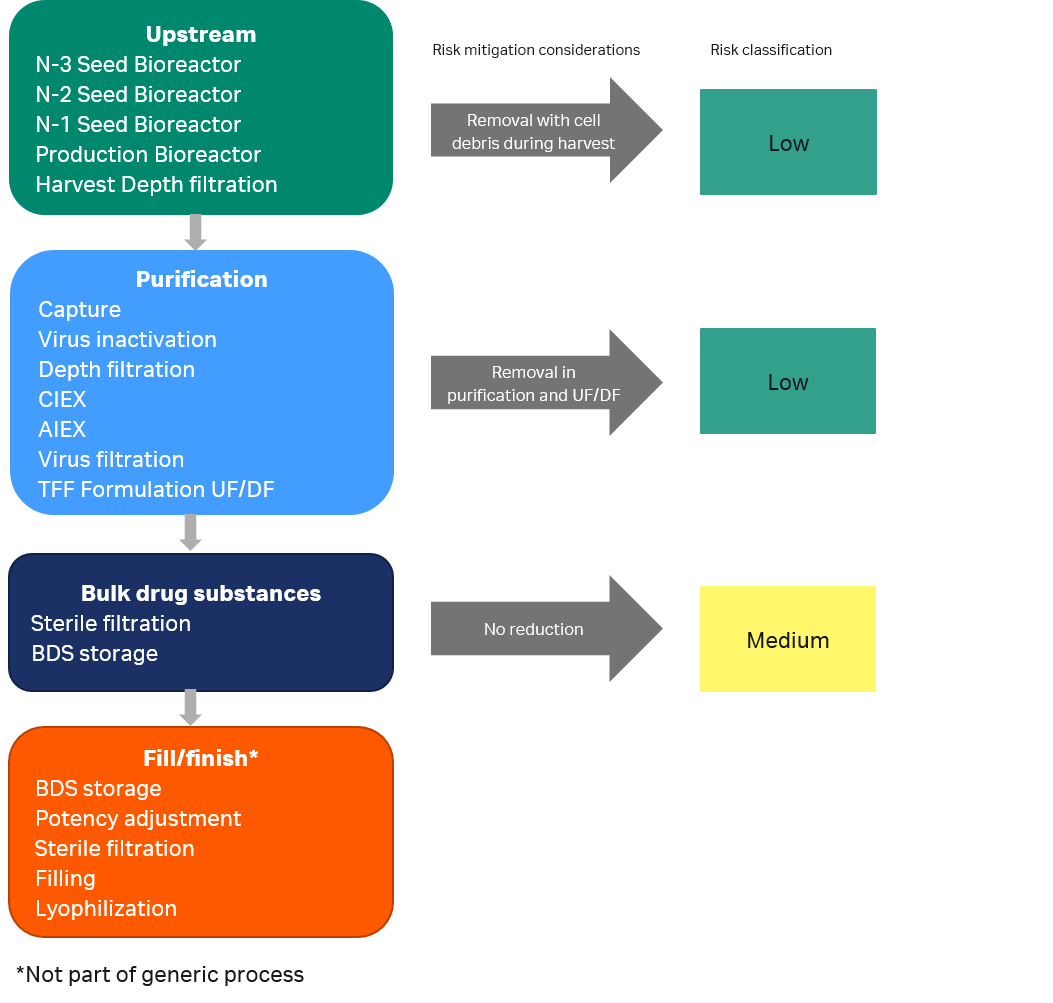
Fig 5. Risk classification of components in different process steps after risk mitigation considerations.
For medium risk consumables, extractables study reports should be collected and evaluated for relevance. Study conditions should be compared with actual process conditions to make sure that these are bracketed. The chemical analysis techniques used in the studies should be reviewed to check the soundness and completeness of the outcome.
Process specific extractables data — a step-by-step approach

In order to properly assess the risk with extractables as potential leachables, it is necessary to know the identities of the substances and the levels to which they could accumulate. As the risk model indicates, consumables used in the late process steps will likely contribute most to leachables. In the generic mAb manufacturing process, any process equipment related leachables entering the fill/finish step with the purified mAb, would most likely be derived from the product hold bag after UF/DF filtration, the pump tubing, the sterilizing grade filter, and the final bag for holding the bulk drug substance (BDS). The following steps were taken to obtain an appropriate set of extractables data for these consumables from results in the extractables study reports. The step-by-step approach is described in the bullet points below.
- Select the extractables data sets which bracket the process conditions, i.e., the relevant exposure duration and model solvents.
- Multiply (extrapolate) the extractables data sets to correspond to the wetted surface area and number of components of the consumable. When results from several solvents are used to bracket a process solution, use the highest result for each extractable compound over all the selected solvents.
- Add the contributions from the different components of the consumables. These are the estimated extractables for the consumable.
- Repeat the procedure for each of the consumables.
Extractables were thus estimated based on results from consumable assemblies, components and materials, to the extent available. The contributions of extractables from the consumables used after the UF/DF step, i.e., the product hold bag, pump tubing, sterile filter, and bulk fill bag, were added together. In addition, 10% of the results on extractables from consumables in the prior formulation step were added. Because of the efficient reduction of extractables demonstrated with UF/DF, 10% was considered a conservative estimate to be used for the contribution. Together, the findings are considered to represent major extractables which may become leachables with the mAb in the final bulk fill bag.
Chemical safety assessment of extractables
Once an appropriate extractables data set is available, a drug product-specific safety assessment based on production batch size and dosing regimen may be conducted to evaluate patient safety aspects of extractable compounds.
Based on a general example, including manufacturing process settings, route of administration, and dosage level the potential extractables from consumables in the described process was evaluated. Assumptions are summarized in Table 4.
Table 4. Assumptions for a generic mAb production process.
| Variables | Assumptions |
|---|---|
| Batch size |
|
| Drug dosing |
|
| Exposure |
|
As described above, extractables from the late unit operations were added together:
- 100% of extractables generated from consumables in sterile filtration and BDS storage
- 10% of extractables generated from consumables in formulation and UF/DF
The extractables data set calculated for consumables in the late process steps was used for the chemical safety assessment. In our case, the data indicated 36 extractable compounds at a level > 1 mg. The highest individual compound was calculated at 141 mg. Using the process assumptions in Table 4, the potential exposure to the most prevalent extractable compound can be calculated:
- Exposure = Extractable/mAb × dosage
- Highest exposure = 141 mg /4.2 kg × 7.14 mg/person-day = 0.24 µg/person-day
Comparison of this potential exposure can be made to a threshold of toxicological concern (TTC) limit set at 1.5 µg/person–day with treatments > 10 years to lifetime (11). This limit is associated with a negligible risk (theoretical excess cancer risk of < 1 in 100,000 over a lifetime of exposure) and can in general be used for most pharmaceuticals as a default to derive an acceptable limit for control. The conservatively calculated potential exposure is found below the TTC limit with the highest individual extractable compound, and consequently with all individual extractables found at a lower level. Hence, the safety risk with extractable compounds was found acceptable.
Leachables study considerations
A leachables study is typically conducted when components have been determined to carry a high risk and therefore may require leachables testing according to the best practices guide (3). Our mAb manufacturing process discussed ends with bulk fill and none of the consumables were classified with high risk, which indicates that a leachables study would not be needed with these process consumables. Also, it was shown with a chemical safety assessment that toxicity risk was found acceptable within the assumptions discussed and does not infer that a leachables study would be needed prior to fill and finish.
There may, however, be other specific reasons for performing a leachables study and this is up to every manufacturer to decide. If so, the generated extractables data should serve as a source of information to what leachable compounds would be expected. With final fill and finish there is generally a higher risk rating and increased demands on performing a leachables study. Adoption of best practices and use of Cytiva extractables data can help manufacturers in risk assessment and in design of leachables studies to ensure product quality and patient safety following implementation of single-use systems in biopharmaceutical manufacturing processes.
Conclusions
A step-by-step procedure on how to assess extractables and leachables derived from a full single use biomanufacturing process, from the seed bioreactor to the final bulk fill storage bag, was presented in this report. The industry best practices for assessment of leachables were applied and available extractables data from Cytiva was used.
Risk mitigation considerations must be applied for an estimation of extractables over a complete manufacturing process, otherwise, estimated extractables may not become representative of potential leachables. First, low risk consumables should be identified and discounted in order to expedite the assessment. Secondly, contributors and detractors of potential leachables in the process should be considered so that focus can be placed on relevant extractables.
The compilation of extractables for the generic mAb process was made from consumables classified with medium risk. After consideration of decreases in the number of extractables in UF/DF and removal of parts of the contributions, major extractables were listed as potential leachables. A chemical safety assessment performed on the results indicated that the extractables generated during the assumed process conditions, ending with the bulk fill, would be below level of concern and therefore leachables testing should not be needed on these consumables.
References
- 21CFR211.65 (a) Equipment construction.
- BioPhorum best practices guide for extractables testing of polymeric single-use components used in biopharmaceutical manufacturing. April 2020.
- BioPhorum Operations Group (BPOG) best practices guide for evaluating leachables risk from polymeric single-use systems used in biopharmaceutical manufacturing.
- K. Paudel, A. Hauk et al., Quantitative Characterization of Leachables Sinks in Biopharmaceutical Downstream Processing. European Journal of Pharmaceutical Sciences. 143:105069, Oct. 2019.
- Armin Hauk et.al., Filtration membranes - Scavengers for leachables? European Journal of Pharmaceutical Sciences. 120, April 2018
- Shukla A. et al., Downstream processing of monoclonal antibodies – application of platform approaches. J. Chromatogr. B. 2007; 848: 28-39
- Van Reis R. et al., Protein ultrafiltration. Encyclopedia of Bioprocess Technology: Fermentation, Biocatalysis, and Bioseparation. Flickinger MC, Drew SW, ets. Wiley, New York, 1999: 2197-2214
- Shao J. et al., Optimization of ultrafiltration/diafiltration processes for partially bound impurities. Biotechnol. Bioeng. 2004; 87: 286-292.
- N. Magarian et al., Clearance of extractables and leachables from single-use technologies via Ultrafiltration/Diafiltration operations. Biotechnol. Prog., 2016; 32 (No. 3): p. 718-724.
- Example of dosage for Enbrel™ is 25 mg two times per week for adults, i.e., 7.14 mg/day.
- Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk, ICH M7 (R1).