The goal of biomanufacturing is to deliver safe, high-quality therapies to patients. For this reason, extractable and leachable analysis is an essential component of any quality program. The regulations and best practices for evaluating extractable and leachables continue to change over time (Fig. 1). It is important to stay up to date to ensure regulatory compliance and ultimately patient safety. From our years of experience developing single-use technologies, Cytiva has extensive knowledge of extractable and leachables testing for bioprocessing materials. This article will provide an overview of the regulations around extractables and leachables for single-use technology.
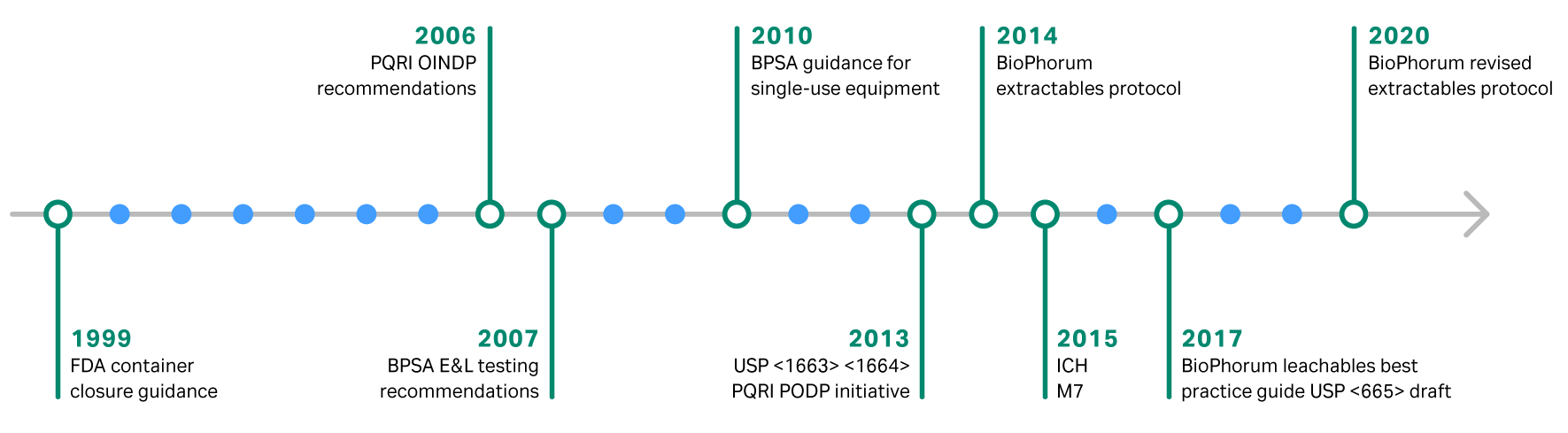
Fig 1. Evolution of extractables and leachables regulations
Standardization of extractable and leachable testing
The adoption of single-use technologies continues to grow due to the benefits they provide, such as faster time to market, increased efficiencies, and support of sustainable and economical processes. With a diverse range of fluid contact materials compared to stainless steel and glass, single-use technology requires more extractables and leachables data to qualify the equipment into your processes.
Industry standardization of extractables and leachables analysis through BioPhorum protocols makes the adoption of single-use products more efficient. Since 2014, this industry association has been working to make extractable and leachable testing as efficient as possible.
In 2014, BioPhorum published a best practice guide for performing extractable testing of single-use components in biopharmaceutical manufacturing. This guide was updated and streamlined in 2020. It provides recommendations on:
- Selection of extraction testing conditions,
- Study designs for a range of single-use components, and
- Recommended analytical methods.
In 2020, United States Pharmacopeia published a new revision of draft chapter USP <665>. USP <665> and its companion guideline USP <1665> provide a framework for assessing the plastic materials used in the manufacturing of drug products. From a single-use perspective, this includes components such as bags, connectors, sensors, valves, and tubing, among others.
Tracing extractables and leachables back to the source
Identification of extractables from the single-use device and traceability of these to the source of the raw materials is key to securing consistent quality and risk mitigation. Table 1 summarizes some of the factors that require consideration when performing an extractables and leachables study.
Table 1. Factors to consider when evaluating extractables and leachables.
| Risk assessment of product contact materials | |
| (1)
Process conditions covered by BioPhorum Extractables and Leachables guidelines* |
Extractables assessment
Identify all product-contact materials, conduct a detailed material assessment using supplier extractable data, or design a custom study per your process needs. |
| (2)
Toxicological evaluation |
Toxicological risk assessment
Assess toxicological risk from extractables by taking into account the dosage form, route, and frequency of drug administration. |
| (3)
Extractables levels acceptable to guidelines |
Leachables study for final container closure
Measure and assess the levels to which leachables will accumulate in your finished drug product over its shelf-life in its final container closure. This information can be used to support your regulatory filing. |
| APPROVED to patient safety standards |
* Consistent with USP <665> and USP <1665>
A thorough risk assessment for extractables and leachables testing requires identifying possible sources specific to the process. The components can then be categorized into levels of risk for extractables and leachables based on their use within the process (Fig. 2). For example, single-use polymeric components that are in the steps closer to the drug formulation carry a higher risk rating for extractables and leachables.
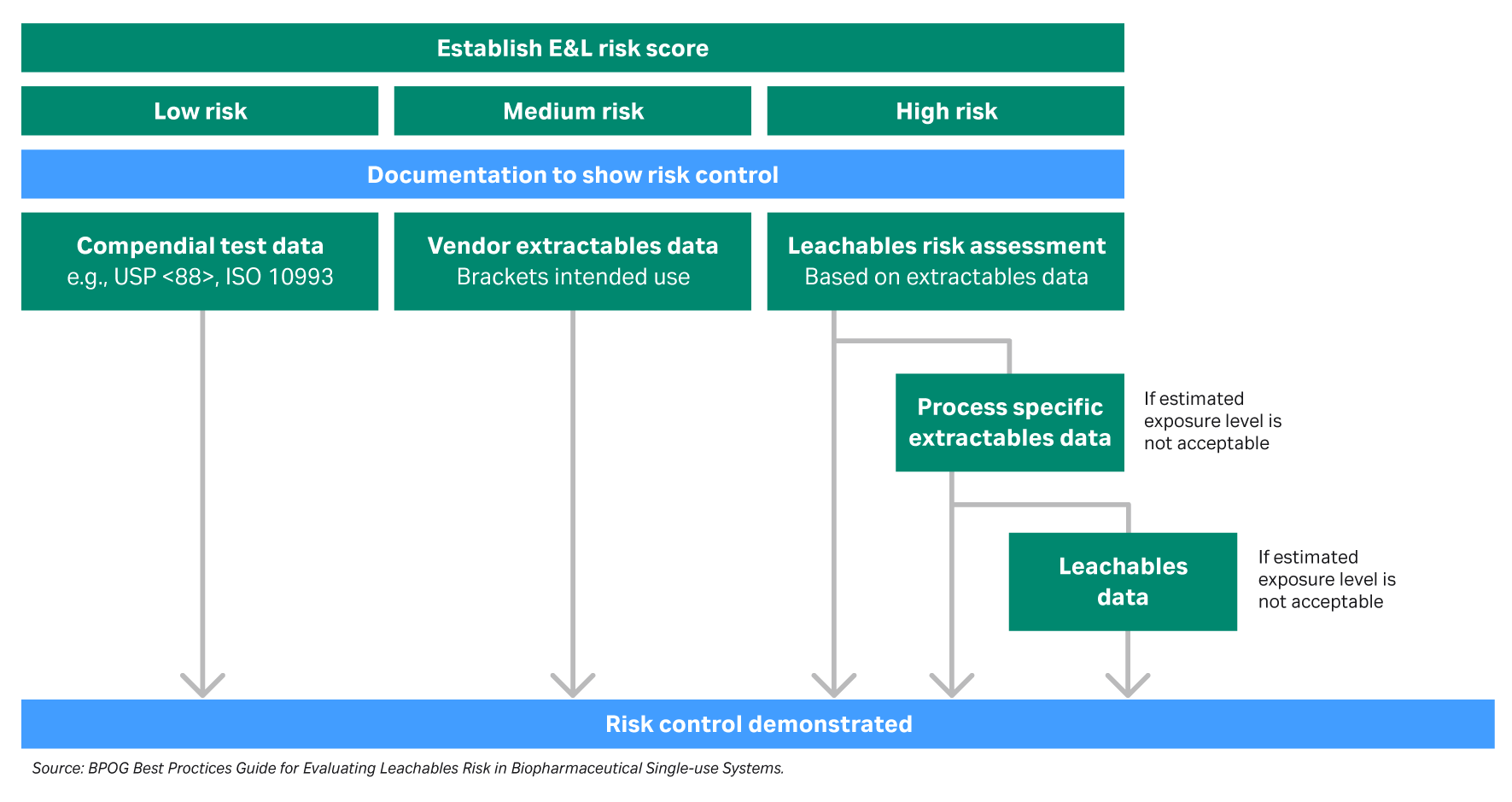
Fig 2. Extractables and leachables risk assessment process flow diagram.
The analytical evaluation threshold (AET) is defined as the threshold at or above which a chemist should begin to identify a particular leachable and/or extractable and report it for potential toxicological assessment. The AET is a relative value based on the safety concern threshold (SCT) and the drug product dosing. The AET is developed during extractables studies and is applied to both extractables and leachables. Targeting an appropriate AET can significantly improve the efficiency of extractables and leachables testing.
Approach to extractables and leachables at different phases of drug development
An extractables risk assessment is required as part of your regulatory filing prior to commercial manufacturing. However, you can choose to perform this assessment in earlier phases of development.
An extractables risk assessment in the discovery phases (prior to Phase III) should consider:
- Materials of construction,
- Compatibility with manufacturing conditions, and
- Extractables data from vendors
This information can be used to establish the magnitude of patient exposure and the overall safety risk posed by an extractable. If the assessment finds that there is an unacceptable level of risk, further testing can be conducted.
A standardized study design will cover a wide range of possible manufacturing and storage conditions. However, there will be times when the process conditions fall outside of a standardized protocol. A custom study is needed in this case. A well-designed study:
- Includes test conditions that bracket actual drug manufacturing conditions
- Includes test items that are representative of the actual drug manufacturing process components
- Applies appropriate pre-treatment such as gamma irradiation, steam sterilization, or flushes/rinses
- Sets an appropriate AET
- Uses orthogonal screening methods
Contact Cytiva for support with custom extractable and leachable studies based on your objectives and risk profile.