What is process development for biopharmaceuticals?
Definitions, activities, and things to consider when developing an upstream or downstream bioprocess.
Biopharma process development comprises the activities that help you create a series of steps to produce a biomolecule – a monoclonal antibody (mAb), recombinant protein, viral vector, or other product that comes from a biological origin.
Bioprocess development is often divided into upstream process development and downstream process development. Those activities must be combined with the right analytics, so you can accurately measure what you've identified as your product's critical quality attributes (CQAs) as you develop and refine your processes.
Process development activities will vary by your type of biomolecule, as well as the stage of the drug development process you are in – preclinical, early clinical (Phase I/Phase II), or late clinical (Phase III/Phase IV). Regulatory requirements will guide many of these activities.
At early stages of drug development, you will develop a process that is 'good enough' to meet the needs of that stage. However, it's important to keep the end goal in mind. Ultimately, you will need a process that translates to a manufacturing environment, one that is easy to scale up throughout clinical trials and to the market. By the time you reach Phase III in the drug development process, 'good enough' is not sufficient. Instead, you will switch your focus to making sure you have a robust upstream or downstream process that delivers a high yield and high productivity. Also, your process must be cost-effective and reproducible.
Keep the end in mind for future success
Key decisions early on will impact how you accomplish your goals. For example, a suspension cell culture is easier to scale up compared to adherent cells. And ultracentrifugation to separate empty from full capsids of adeno-associated virus (AAV) does not scale well. Consider scalability from the start to avoid the need to rework your process.
And if you make changes to your manufacturing process during clinical trials, this could impact regulatory CMC (chemistry, manufacturing, and controls) submissions and could have a negative effect on timelines further down the road. So, if you're looking for long-term success, it's important to take the time early on to get the process design right.
This is especially true if you're working on a biomolecule and indication that regulators might grant an accelerated pathway to approval. By making key decisions early around process development, quality assurance, and quality control strategies, you can set yourself up for success in current good manufacturing practices (cGMP) manufacturing. Focus your efforts on making sure that your manufacturing process is robust.
Process development must balance product quality with process performance (Fig 1). Market needs for speed and economy are driving the need to make go/no go decisions earlier, as well as to bring products to market faster and at a better price point. These needs translate into growing pressure to quickly create processes that are cost-effective at manufacturing scale. When added together, gains in productivity can have a big positive impact on process economy.
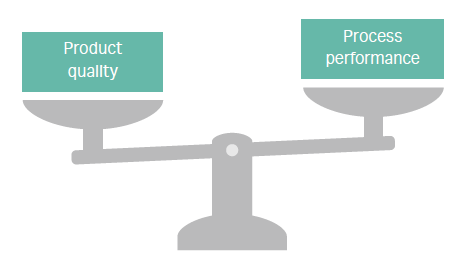
Fig 1. Biopharma process development is a balancing act.
Are you curious about CMC, cGMP, and other key terms? Check out our definitions for bioprocess development.
Process development activities by stage of drug development
The extent of process development will vary by the stage of the drug development process. The general progression of the phases is similar around the world, but the specifics will vary by region depending on guidelines from the relevant regulatory authority.
Process development for tox and early clinical stages: focus on what’s important
In these early stages, your process development efforts are key to ensuring a robust product over time. Here, you will focus on a few important parameters. Your efforts must support manufacturing of the product for toxicology (tox) and Phase I/II studies and support your regulatory filings. Your goal is to develop a process with intermediate yield while meeting all product quality expectations. This first version of your process is developed with the initial product quality specification. The developed process should have the potential to scale to full-scale manufacturing without much modification.
To focus your process development activities on the key process parameters, you need to identify these parameters in a structured exercise that takes prior knowledge and experiences into account and helps to prioritize experiments. You can envision process parameters using a fishbone diagram. To prioritize activities, you can use a prioritization matrix or a failure mode and effects analysis (FMEA).
At the end of your downstream process, you will have the drug substance (DS). This will be formulated into the drug product (DP). Be sure to start pre-formulation activities early. Keep the DP formulation simple at this stage; perhaps formulate it as a frozen solution.
Process development for Phase III and commercial manufacturing: make sure your process is robust
At this stage, you must have a process that is suitable for commercial manufacturing. Process development work is extensive and typically uses design of experiments (DoE) studies. Here you will focus on defining your design space using quality by design (QbD) principles. You will optimize and perform extensive work on critical process parameters (CPPs) through process characterization. Overall, you will focus on robustness, yield, reproducibility, and scalability, ultimately generating a cost-efficient process.
At the late clinical stages you will develop the DP formulation for market launch. For example, you might consider lyophilization to extend the shelf life. For your regulatory filings you will need to demonstrate comparability between the Phase I and Phase III product. The reason for this is to show regulators that the product didn’t change over time and that clinical trial results are representative and supportive of the commercial product.
Refer to the following sections for a detailed breakdown of early- and late-stage activities:
Upstream process development
Downstream process development
Are you curious about IND, CMC, and other key terms? Check out our definitions for bioprocess development.
Upstream process development: fundamentals
Upstream process development comprises the workflow from cell line development to a developed bioreactor process prior to the harvest and purification steps. Cell line development (CLD) is a critical activity in biologic manufacturing processes (exception: in autologous cell therapy, where the starting material comes from the patient’s own cells). Advances in CLD have helped to increase titer and allow process developers to move molecules into the clinic faster. Different therapeutics might use different host cell lines, but in all cases a single clone is required for a biopharmaceutical process.
The production system will depend on the type of biomolecule you are interested in. For example, if you are producing an antibody fragment that doesn’t require a specific post-translational modification (e.g., glycosylation pattern), you might choose a microbial system. On the other hand, if your product must be made in a mammalian system, there are many specific cell lines you can use based on Chinese hamster ovary (CHO), human embryonic kidney (HEK), or other mammalian cells. Insect cells are suitable for producing some biomolecules. Developing the cell culture medium platform is another important step in upstream process development. For any process, the ideal scenario from a regulatory point of view is to use media and feeds that don’t have any animal-derived components (i.e., are ADCF), as it lowers the potential risk of animal-derived adventitious viruses.
Specific activities in upstream process development include:
- cell line development (host cell line engineering, transfection, clone selection, stability testing)
- media and feed optimization
- optimization of operating parameters (pH, temperature shift, gassing strategy)
- scale-up strategies in bioreactors or fermentors.
Before you start Phase I/Phase II
Early-stage upstream process development starts with generating a specific cell line and selecting a single clone. Here you will assess the growth, stability, and productivity of your production system in bench-scale experiments.
It's important to include the time to produce good manufacturing practices (GMP) cell banks prior to the toxicology (tox) and Phase I/Phase II campaign. Make sure you have freedom-to-operate or commercial licenses in place for the parental cell line and vectors used.
Before you start Phase III
Late-stage upstream process development is focused on defining and optimizing the key parameters to improve the productivity, process robustness, and product quality. To achieve consistent production of a biologic, you must maintain your ranges of critical process parameters (CPPs) and critical quality attributes (CQAs) throughout the process. To do so, you must know your CQAs and identify the CPPs and their ranges and understand the risk associated with the process. You must also develop risk mitigation strategies for commercial production. The design space defines the acceptable ranges for the CPPs to meet the product quality specifications, and this is a key aspect of the quality by design (QbD) tool.
Table 1. Summary of upstream process development activities by clinical stages
| Upstream development stage | Activities |
| Early clinical stages |
|
| Late clinical stages |
|
Reference
Adapted table used with kind permission: Lindskog E., “Chapter 31 The upstream process: principal modes of operation” in G Jagschies et al., “Biopharmaceutical Processing: Development, Design, and Implementation of Manufacturing Processes”. Elsevier 2018.
Are you curious about CPP, CQA, and other key terms? Check out our definitions for bioprocess development.
Downstream process development: fundamentals
A downstream process refers to the recovery and purification of the target molecule after the upstream process. It includes harvest, purification, and viral clearance steps. Different and multiple steps are usually arranged and/or combined to purify the product of interest. Tools and process conditions (i.e., the critical process parameters, or CPPs) are defined during downstream process development. The objective is to find the appropriate balance between yield and quality (i.e., the critical quality attributes [CQAs]).
To start out, you will likely perform your process development work at lab scale. To speed up process development and make it more efficient, you can use high-throughput process development (HTPD) for the initial screening of process conditions. Or you can use HTPD for a more thorough investigation of a defined space as a basis for detailed process understanding and/or robustness studies. After scouting and screening of a chromatography process, you will perform verification and optimization with larger columns. This table summarizes the activities by stage of downstream process development.
Table 2. Summary of downstream process development activities by clinical stages
| Downstream development stage | Activities |
| Early stage clinical |
|
| Late stage clinical |
|
Are you curious about CPP, HTPD, and other key terms? Check out our definitions for bioprocess development.
Analytics: why they’re important for process development
Quality attributes are determined and controlled at several key stages of process development and at key points in the manufacturing process. Indeed, critical quality attributes (CQAs) are key to regulatory compliance. A CQA is defined in ICH Q8 as the property or characteristic that defines the quality of the product (i.e., what is critical for the patient.) Regulators also need information on data for CQAs in the process to ensure that process steps work as intended. You need to start with CQAs that characterize the biological function of the molecule. Examples include receptor binding affinities and correct glycosylation. In general, biologics ― viruses, mAbs, cells (for cell therapies), antibody-drug conjugates (ADCs), viruses, and more ― are large, complex molecules that require robust and reliable methods to analyze and characterize them in depth.
Here’s a list of typical analyses for protein-based biomolecules. Other types of biotherapeutics often share some of these analyses but have some of their own.
- Purity
- Identity
- Bioactivity
- Binding activity
- Host cell protein (HCP) and other impurities
- Concentration
- Size and aggregation
- Comparability
Learn more about bioanalytics.
Are you curious about CQA and other key terms? Check out our definitions for bioprocess development.
Definitions for biopharma process development
- What is a biologics license application (BLA)? A biologics license application (BLA) is a formal request to the Food and Drug Administration (FDA) asking for approval to sell and market a new biological (biologic) drug in the United States. A BLA is similar to a new drug application (NDA) but can be much more complex due to the nature of biologic therapeutic manufacturing.
- What is CFR in pharma? CFR refers to the Code of Federal Regulations, which codifies the rules (laws) published in the United States Federal Register. The portion that governs food and drugs is Title 21, referred to as 21 CFR.
- What is cGMP? In the United States, current good manufacturing practices (cGMP) is a set of regulatory requirements that provides for the minimum methodology and controls to produce a safe and effective drug or biologic product. The ‘c’ is used to emphasize that the systems and controls used must be up to date with the latest regulator expectations.
- What is a critical material attribute (CMA)? A critical material attribute (CMA) is a property or characteristic of an input (raw) material that should be kept in an appropriate range to ensure the desired product quality. A CMA can be physical, chemical, biological, or microbiological. Example: purity/impurity levels in iron carriers used to make cell culture media.
- What is CMC in drug development? Chemistry, manufacturing, and controls (CMC) is the section of a regulatory application in USA with details on manufacturing processes, how to ensure product quality, and testing methods. These details give regulators confidence that the product can be manufactured to the same high quality each time.
- What is a critical process parameter (CPP)? A critical process parameter (CPP) is a characteristic of equipment or of a process related to manufacturing, whose variability has an impact on a critical quality attribute (CQA). CPPs should be monitored or controlled to ensure the process produces the desired product quality. Example: factors that impact oxygen transfer to cells in bioreactors.
- What is a critical quality attribute (CQA)? A critical quality attribute (CQA) is a property or characteristic of a product that has an important impact on the therapy’s product quality. A CQA, which can be physical, chemical, biological, or microbiological, must be defined, measured, and controlled to ensure the desired product quality. Example: purity (CD34+) of a chimeric antigen T (CAR T) cell therapy.
- What is CTA? A clinical trial application (CTA) is a generic term referring to the initial submission to permit use of an investigational drug in a clinical setting. The documentation submitted within a CTA is an investigational new drug (IND) application in the United States, and an investigational medicinal product dossier (IMPD) in the European Union.
- What is design of experiments (DoE)? Design of experiments (DoE) is a systematic way to vary inputs to a process and analyzing the results for a cause-effect relationship. The DoE technique minimizes the number of experiments by varying several parameters at the same time.
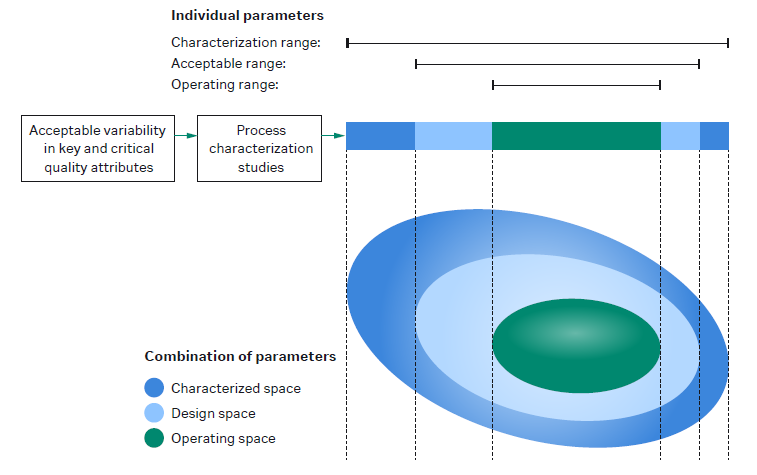
For more information: View short video on understanding DoE in protein purification. - What is design space in QbD? Design space in quality by design (QbD) is the combined interactions of the input materials and process parameters that have been shown to assure product quality. The design space is a subset of the characterization range, which is all the knowledge obtained during process development. Within the design space, a smaller control, or operating space, is defined.
The figure shows the relationship between characterized space, design space, and operating space. - What is the difference between drug substance and drug product? The drug substance (DS) is the bulk substance at the end of the downstream process. The DS is formulated into the final buffer and transferred into the final container (e.g., a vial) in the fill/finish step. This is now the drug product (DP).
- What is the EMA? The European Medicines Agency (EMA) is responsible for the scientific evaluation, supervision, and safety monitoring of medicines in the European Union.
- What is the FDA? The Federal Drug Administration (FDA) is a United States agency responsible for ensuring that drugs, vaccines, and other biological products and medical devices intended for human use are safe and effective.
- What is a first-in-human (FIH) study? A first-in-human (FIH) study is the same as a Phase I clinical study. A first-in-human study is done in people, in contrast to in vitro or animal preclinical studies.
- What is good laboratory practice (GLP)? Good laboratory practices (GLP) describes the regulatory requirements for a quality system for nonclinical safety studies (i.e., preclinical safety studies in animals). In general, the methodology of planning, performing, monitoring, recording, archiving, and reporting is also applied to process development and is often referred to as GLP.
- What is high-throughput process development (HTPD)? High-throughput process development (HTPD) is a way to speed up process development. Typically, HTPD benefits from automation and running multiple conditions in parallel. A wide range of experimental conditions can be evaluated at the same time instead of waiting for the results from one experiment before starting another. HTPD can be used to characterize the design space and helps to define the process parameters that need to be monitored and controlled. The high number of experiments resulting from HTPD creates large data sets. It’s important to have high-throughput analytics in place to analyze these.
- What is the ICH? The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) harmonizes regulatory guidelines that are applied by a growing number of regulatory authorities around the world.
- What is an IMPD? An Investigational Medicinal Product Dossier (IMPD) is required whenever a clinical trial is intended in one or more member states of the European Union.
- What is an investigational new drug application (IND)? An investigational new drug application (IND) is a request for the US Food and Drug Administration (FDA) to authorize giving an investigational new drug or biologics to humans. An IND is required before starting a Phase I clinical trial in the United States and is updated for the subsequent clinical stages. All regions require some type of application in order to conduct human clinical studies. The application name may be different in the various regions (i.e., IMPD in Europe, CTA in Canada, IND in China, etc.).
- What is an MMA? A Marketing Authorisation Application (MAA) requests permission from the European Medicines Agency to introduce a new drug to the market in the European Union.
- What is the difference between BLA and NDA? Both are formal requests to the US FDA asking for approval to sell and market a new drug in the United States. A BLA or biologics license application, applies to a drug that’s a biological product. An NDA, or new drug application, applies to all other drug products (i.e., a small molecule drug.
- What is an NDS? A New Drug Submission (NDS) is a generic term for applications to request permission to introduce a new drug to the market.
- What is the NMPA? Formerly known as the China Food and Drug Administration or CFDA, the National Medical Products Administration (NMPA) is the Chinese regulatory authority for drugs, medical devices, and cosmetics.
- What is a pharmacokinetics/pharmacodynamics (PK/PD) study? A PK/PD study analyzes the drug concentration in the body over time, how it affects the body, and how it clears the body.
- What is a platform in biomanufacturing? In this context, a platform is a specific set of equipment or a specific process for manufacturing related molecules. Instead of starting from scratch with each new molecule, a platform builds upon knowledge and experience gained in an iterative way. Ideally, a platform will require only slight changes to adapt it for related molecules, which should save time and costs.
Example: a range of bioreactors that lets you scale up or down as batch sizes change. - What is a process analytical technology (PAT)? Process analytical technology (PAT) is a way to measure, analyze, and control critical process parameters (CPPs) that affect critical quality attributes (CQAs) of a product. PAT focuses on timely measurement, preferably during a process, and is a key component of using a quality by design (QbD) approach to biopharmaceutical manufacturing. Example: chemical optical sensors for bioprocess monitoring to ensure CQAs are met.
- What is process characterization? Process characterization studies provide a deep understanding of the purpose of each process step and the effect of process inputs on the process outputs. Process characterization studies can help when investigating process deviations.
- What is process robustness? Process robustness is the ability of a process to tolerate defined sources of variation, such as variation in input materials or changes to process equipment, without impacting product quality.
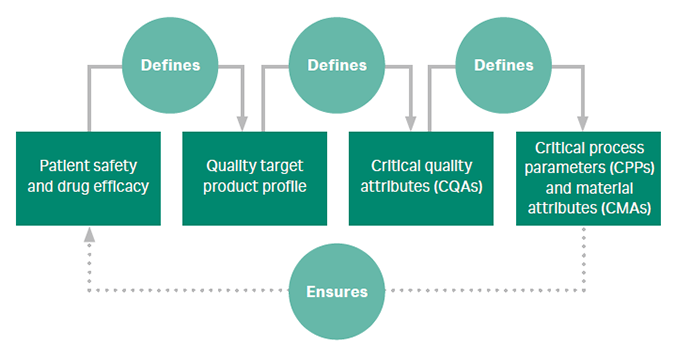
- What is quality by design (QbD)? Quality by design (QbD) is a set of principles that guide design of products and processes with the idea that quality can be ‘built in’ from the start. QbD is an important process development tool to define parameters that ensure quality, yield, and a robust process that tolerates variability.
The figure summarizes the systematic approach to QbD. - Screening design of experiment (DoE) identifies which of many factors or conditions used have a significant effect on the response. Example: screening to predict step elution conditions for a cation exchanger.
Resources
U.S. Department of Health and Human Services, Food and Drug Administration. Guidance for Industry PAT — A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance. Sept. 2004. Accessed July 18, 2021.
International Conference on Harmonization (ICH) and FDA Guidance for Industry, Q8 (R2) Pharmaceutical Development, Nov. 2009. Accessed February 22, 2021.
European Medicines Agency. Quality by design. https://www.ema.europa.eu/en/human-regulatory/research-development/quality-design. Accessed February 22, 2021.
Regulatory: What to think about during process development
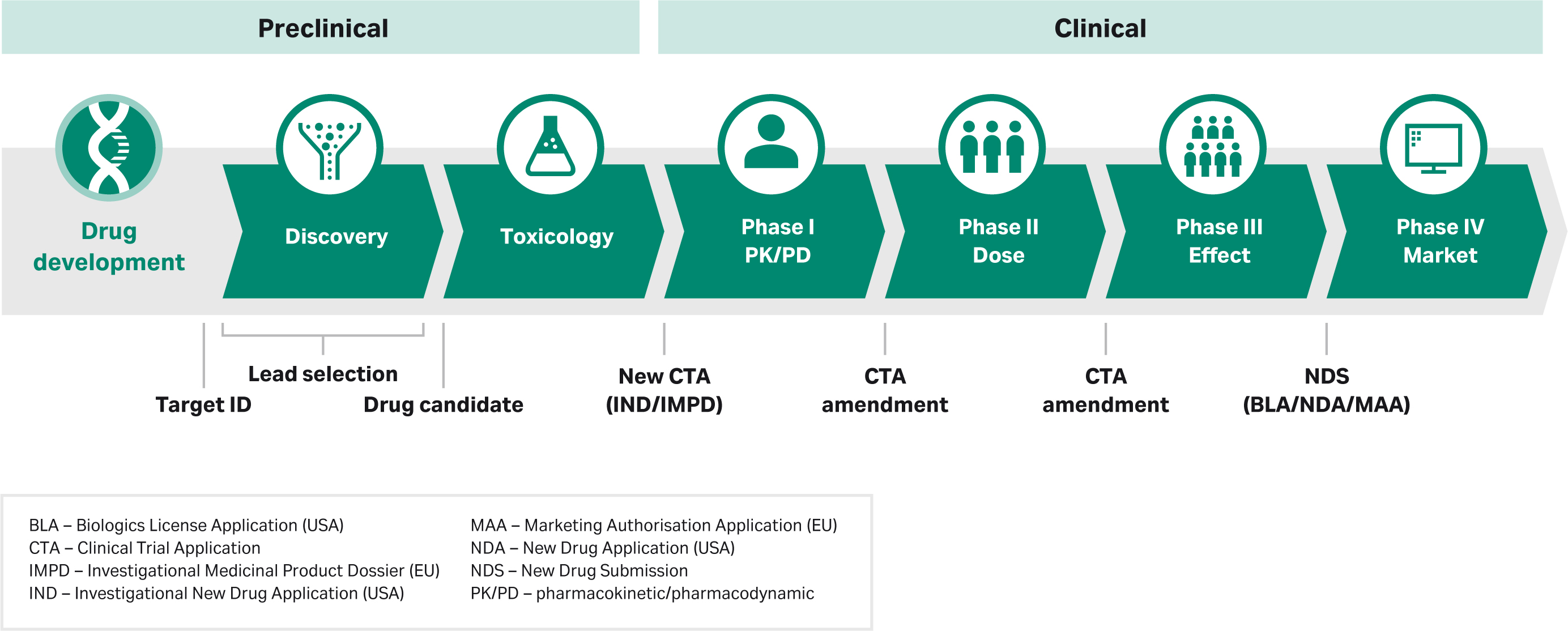
What happens during process development has a big impact on meeting regulatory requirements. In this section we highlight the process in the United States. The general progression of the phases is similar elsewhere in the world, but the specifics will vary by region depending on guidelines from the relevant regulatory authority, such as the European Medicines Agency (EMA) in Europe or the National Medical Products Administration (NMPA) in China. Here’s a diagram showing where regulatory filings for the US FDA come into play during the drug development process with some terminology added to correlate to other regions:
Pharmacology and toxicology data from in vitro and animal studies are essential before moving on to clinical trials. This data is required to submit an investigational new drug (IND) application. The IND application represents a pivotal milestone, as the data that’s included is reviewed by regulators to determine if there is sufficient justification for a Phase 1, a first-in-human (FIH), trial, or a Phase 1 new therapeutic indication of use. The small Phase 1 studies confirm drug candidate safety for further investigation.
While IND applications need to include basic product characterization data for Phase 1 protocol clearance, the information included in the IND builds continually over time. The document will follow you through the regulatory approval process, expanding to include data from all studies done from phase to phase and in parallel. The IND application will include a section on chemistry, manufacturing, and controls (CMC), which will also expand throughout clinical trials as more data and process knowledge is gained over time.
The steady development of information included in the IND is critical, as regulators use this data to determine a drug’s eligibility to advance through the approval process. For example, you might not finalize formulation, route of administration, indication, and dosing specifics until several options are tested in exploratory Phase 2 studies with affected patients. Once these key metrics are defined, the data supporting final formulation, dosing, target population, and indication is added to the IND. With each round of regulatory reporting, regulators review new information and, if deemed suitable, clear the candidate drug for larger studies, such as confirmatory Phase 3 trials.
For some drug candidates, regulatory agencies can grant an accelerated pathway to approval. In those instances clinical phases can be combined to accelerate timelines. But regulators must still be convinced that the drug’s safety and efficacy is where it should be.
For clinical trials in China, NMPA suggests having some flexibility on Phase 1, 2, and 3 design, and can combine phase 1 / 2 together as early clinical trial phase and phase 3 as confirmatory clinical trial phase. This is followed by phase 4, the post-market phase. NMPA also recommends following ICH guidelines wherever applicable. NMPA has published several guidance documents for cell and gene therapy – see the Resources section.
As in other aspects of drug development, up-front planning for regulatory can prevent headaches down the road. One of the key decisions is knowing when your team is short on regulatory experience and expertise, and when to call on a consultant or hire an expert to fill the gap.
To avoid surprises, don’t wait until you do your filing to engage with a regulatory agency. There are several meetings that can be arranged to gain regulator feedback, such as a pre-IND meeting with FDA. Seek their advice ahead of time according to their defined procedures.
Resources for cell and gene therapy products in China
National Medical Products Administration. Technical guidelines for research and evaluation of cell therapy products, 2017.
National Medical Products Administration. GMP appendix – cell therapy products, 2019.
National Medical Products Administration. Guiding principles for pharmaceutical research and evaluation of gene therapy products, 2020.
National Medical Products Administration. Technical guidelines for clinical trials of immune cell therapy products, 2020.
National Medical Products Administration. Technical guidelines for clinical trials of human stem cells and their derived cell therapeutic products, 2020.
National Medical Products Administration. Guiding principles for clinical trial design of oncolytic viruses, 2020.
Are you curious about IND, CMC, and other key terms? Check out our definitions for bioprocess development.