Article inspired by a Tapas and TECH Talks digital event with insights from customer collaborations.
By leveraging early decisions around process development, quality assurance, and quality control strategies, you can set yourself up for a successful cGMP manufacturing workflow. This article will tackle how to approach process development with a manufacturing workflow mindset that will transition smoothly to cGMP manufacturing.
Understanding robust design
Directed process development means that you focus your efforts on ensuring that your manufacturing process is robust. To understand robust design, you must know the stage of process development when things will start to have a major impact. From a compliance perspective, we have a saying where ‘the process is the product’. So, key decisions early on will impact how you are going to accomplish your goals.
Key questions:
- Will you use a manual or functionally closed process?
- Will you go with single-use?
- Are you using a dedicated equipment train?
Several factors can play in here, and the big three are:
- How fast can you get the project going?
- How much will this project cost?
- How effective will these choices be?
It would be ideal to achieve high speed, low cost, and high quality (Fig 1). Normally, you can only have two out of the three options.
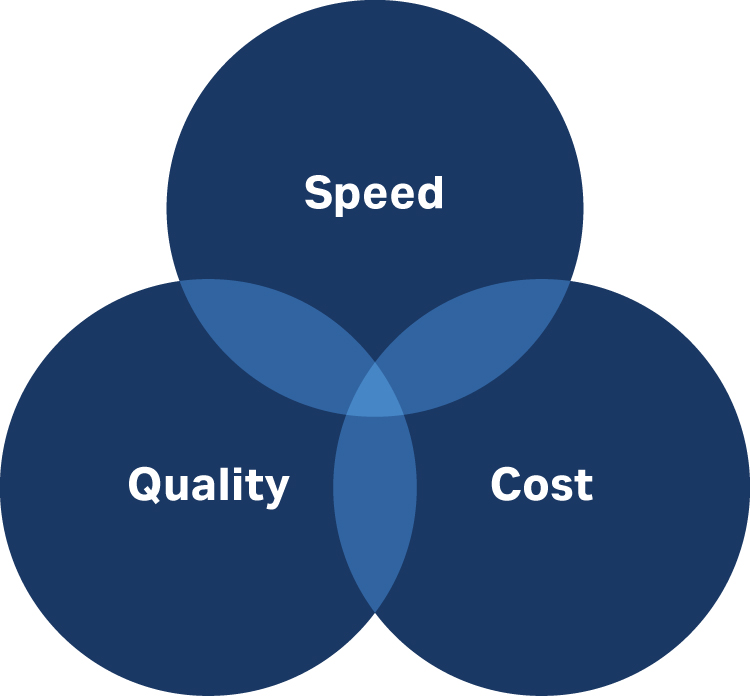
Fig 1. When developing a cell therapy process, consider how you will balance quality, speed, and cost.
But most importantly, only quality counts. If you do it right the first time, you avoid the time and cost involved in doing it right the second or third time.
Managing variation
Robust design is about managing variation. Start with knowing your sources of variability. The basic categories of variation are shown in Figure 2. The circle sizes represent their relative contributions.
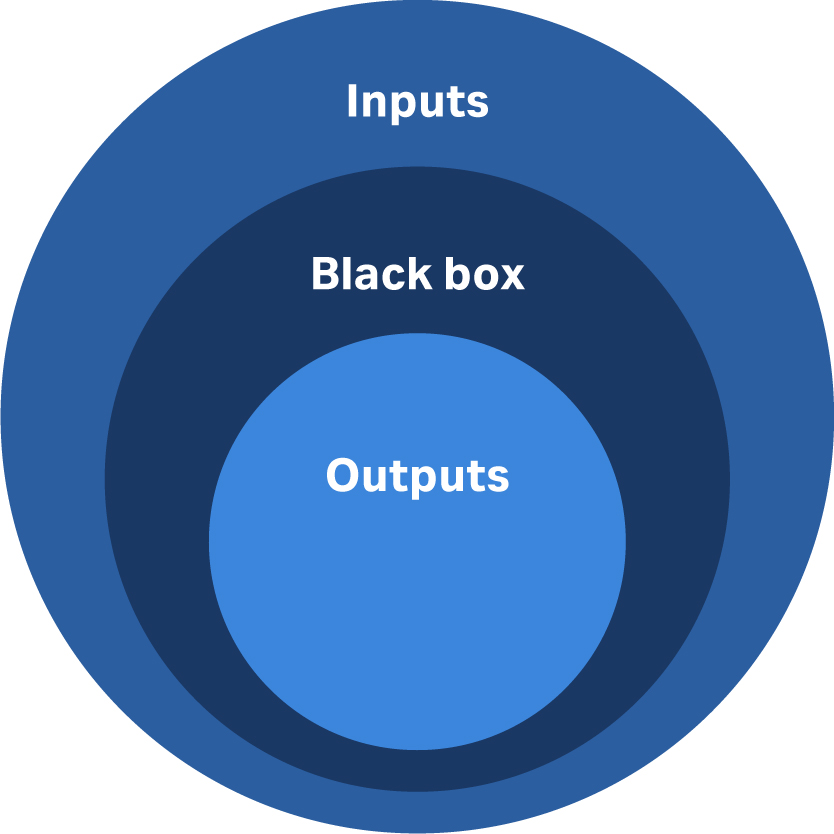
Fig 2. The main categories for sources of variability in a cell or gene therapy process.
The biggest impact comes from variability in inputs, which include raw materials, labor, choice of equipment train, and analytics.
The next biggest impact is what’s going on during the run itself, which is sometimes called the ‘black box’. This variability is dependent upon how well the facility and equipment are running, which is dependent upon the auxiliary support, labor, and how experienced or well-trained the labor force is. Lastly, the outputs are normally the smallest variable. However, at CCRM we find that there can be significant variability in the volume of product manufactured. Typical manufacturers rely on a measure of performance that compares the output against what’s considered a theoretical yield. But with cell and gene therapy, the theoretical yield is very challenging to calculate. Also, there can be variations in shipping, specifically cold chain concerns.
Process capability
This is a mathematical measure of the levels of variability a system can handle, yet still provide quality outputs. And this is truly a measure of robustness. There is variability not only in the processes inside the flasks or bioreactors, but also in all the processes involved in manufacturing. These processes include the measures of performance, which use the analytical methods to monitor the quality and process performance in the process variability description.
To understand robust design, imagine a process that is relatively in control but still has natural variations. As manufacturing scientists, we will then work on refining the process to further reduce this natural variability. These efforts are considered Six Sigma applications. For every process, several analytical methods are used to help monitor the progress. And each of these methods will have its own natural variability. However, ideally you want the magnitude of variability in analytical methods to be much less than the natural variability in the process in order to detect, with significant resolution, the variations in the manufacturing process.
One risk to doing very significant process development is that if one makes progress in advancing the manufacturing process by revolutionary change, we may see a shift where the variability could be higher in the analytics than in the process itself. In that case, you would lose the resolution to be able to monitor the process itself. And then, the next challenge is to advance the analytics, as without this resolution in the analytics, the process variability may very well go undetected. When looking at the overall product development lifecycle, CCRM operates in what’s considered the process development sweet spot. Our ultimate goal is to support manufacturing for Phase I and Phase II clinical trials.
In the cell and gene therapy world, technical strategies are often highly variable, because the manufacturing processes are still quite manual. However, manufacturing projects are viewed through a different lens than what is used in a research and development or process development lab. It is not necessary to look for the best performance for individual unit operations. Instead, overall performance and performance robustness are key.
Tip: It is better to have a process that is 70% efficient over 99% of the times it was run, rather than a process that is 99% efficient only 70% of the time. So, it is best practice to send a process back for more process development work in order to do efficiency optimization. This should be done as early as possible, in order to take advantage of the low overall process switching costs.
Control strategies
Tip: Look at closing process systems before entering Phase II. The manufacturing scientists at CCRM look at projects with a goal of progressing the control strategy. The six elements of the control strategy are listed in Table 1, each with a distinct path of progress.
Table 1. Control strategies for understanding robust design
| Control element | Description |
|---|---|
| Process controls | Protocol → SOP → Engineering Batch Record → GMP Batch Record Protocol → Developed Test method (TM) → Qualified TM → Validated TM |
| Material controls |
Materials selected → Materials qualified → USP <1043> |
| Equipment controls |
Equipment selected → Equipment installed → Qualified equipment (IQ/OQ/PQ) |
| Strategic decisions |
Open vs closed, type of bioreactor, type of purification and selection strategy, analytical methods |
| Human controls |
Documented training of personnel from general to specific |
| Facility controls |
Process development space → Pilot plant → Controlled manufacturing space |
Processes start out with an idea on the back of a napkin. CCRM can take that napkin, develop it into a standard operating procedure (SOP), which can then undergo more diligence through feasibility studies. After successfully executing feasibility, we can then get ready to move into pilot plant, or straight into manufacturing areas, and undergo engineering work under an engineering batch record. The engineering batch record can then communicate progress to its full master batch record. Analytical methods start off with a very similar pathway.
Tip: From your material control, select materials, test them under feasibility, and make sure you qualify them before you start your engineering runs.
Tip: For equipment control, select the right equipment, get that equipment installed and in place, and proceed to qualification through IQ/OQ/PQ activities.
Strategic decisions are hopefully made very early on:
- Are you going with an open or a closed system?
- What type of bioreactor system are you going to use?
- What type of purification and collection strategy will you use?
- Which analytical methods do you need to measure performance?
For employee controls, it is important to know the trainings, which must be documented, needed to support a manufacturing lab at a certain stage of development. Facility controls are generally quite fixed. Process development work will be done in a process development space. If an employee has access to a pilot plant, they might have a mid-level access to the process controls. Full-access controls will be in place if employees are working in a manufacturing space. Decisions on control strategies must be timed appropriately in order to progress to the next stage in process development.
When you are performing manual processes in the R&D space, the system is manageable, because you are manufacturing one dose at a time, potentially. But when you get into manufacturing for Phase I clinical studies, you might be making three to five, maybe ten doses. And for a Phase II study, you might make up to 50 doses. The strategy is exponentially more challenging, and more importantly, more expensive as you increase the number of doses. But it is important to think this through early on so you can set up a product for success on the commercial market, where you could imagine manufacturing tens of thousands of doses a year.